What chemical compounds might be present in drinking water?
Use 'Print preview' to check the number of pages and printer settings.
Print functionality varies between browsers.
Printable page generated Wednesday, 8 May 2024, 7:44 PM
What chemical compounds might be present in drinking water?
Introduction
This free course focuses on the chemistry of the p-block elements in Groups 13 to 18 of the Periodic Table. The periodicity in the chemistry of these elements will become apparent and any remarkable effects, such as the so-called inert pair effect, will be highlighted.
The approach adopted illustrates how main-Group chemistry is important in everyday life. This course covers the water that you drink. For instance, a high level of certain anions in water can cause environmental pollution and health problems and are therefore regulated by several EU Directives. Cations are also important, for example calcium salts contribute to the hardness of water and water treatment uses aluminium compounds.
This OpenLearn course is an adapted extract from the Open University course S215 Chemistry: essential concepts.
Learning outcomes
After studying this course, you should be able to:
understand that in drinking water anions have a beneficial range of concentration above which they may have an adverse effect on either human health or the environment. Toxic elements and compounds often block essential biological processes
explain how excess anions or cations in aqueous solution can be removed by ion-exchange chromatography or an ion-exchange membrane
explain how cations such as aluminium can be amphoteric, that is they can neutralise both acidic and alkaline solutions. Also recognise how this enables aluminium(III) ions to be used in water treatment.
1 Water
To flourish on the Earth, human, animal and plant life require a constant source of clean water. In many countries around the world new chemical technology is now needed to ensure economical and sustainable water management. As the population increases, industries develop and the understanding of the health implications of pollutants grows. Pollution is defined as the introduction into the environment (water, air or land) of contaminants, the quantities, characteristics and duration of which are likely to be injurious to human, animal or plant life.
The following questions about water quality arise for chemists:
- What chemicals are dissolved in water, such as anions, cations and organic compounds?
- What levels are considered as safe?
- What levels lead to pollution?
- How are unwanted chemicals and microorganisms removed?
In the following sections, by taking water purification as an example, you will see a number of important examples of where main-Group chemistry is important to our everyday lives. Here, several important anions and cations found in water will be considered. Additionally you will explore some chemistry associated with the nitrogen cycle, phosphororus, aluminium and thallium.
1.1 The water cycle and drinking water
Water moves around the Earth in the water cycle (Figure 1). Natural water on Earth is not completely pure as compounds may dissolve at several points in the cycle and this may lead to pollution if above accepted guidelines.
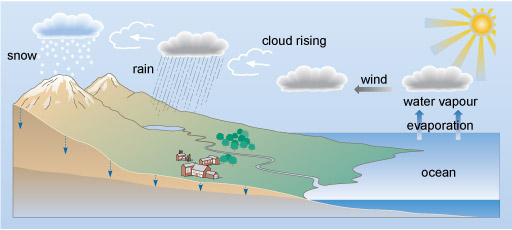
Predict where inorganic pollutants might enter the water cycle?
At several steps in the water cycle inorganic compounds can become dissolved and, depending on their level, pollution may occur. For example:
- Anions such as nitrate and phosphate may become dissolved from agricultural fertilisers, sewage and the natural breakdown of organic matter.
- Air pollution from burning fuels can produce nitrogen oxides, NOx compounds and sulfur oxides, SOx compounds, to form dissolved nitrates and sulfates respectively.
- Cations such as calcium or magnesium may dissolve naturally due to the weathering of minerals in rocks or via anthropogenic, or human-derived, contamination of ground water by pollutants from industry, roads, or mining.
For drinking purposes water has to meet government guidelines but will not be absolutely pure as it is also a natural source of ions needed by the human body. Some typical values are shown in Figure 2 and Table 1.

What will the [H+(aq)] be in the water in Figure 1?
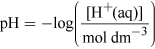 (Equation 1)
(Equation 1)Rearranging gives
[H+(aq)] = 10−pH mol dm−3 = 10−7.4 = 4.0 × 10−8 mol dm−3
| Ion | Concentration/mg l−1 | ||||
|---|---|---|---|---|---|
| Volvic® | Vittel® | Buxton® | Evian® | Tap water* | |
| calcium Ca2+ | 11.5 | 91 | 55 | 78 | 102 |
| magnesium Mg2+ | 8.0 | 19.9 | 19 | 24 | 8.81 |
| sodium Na+ | 11.6 | 7.3 | 24 | 5 | 49.1 |
| potassium K+ | 6.2 | - | 1 | 1 | n.a. |
| choride Cl− | 13.5 | - | 37 | 4.5 | 73.9 |
| nitrate NO3− | 6.3 | 0.6 | 3.5 | 20.6 | |
| sulfate SO42− | 8.1 | 105 | 13 | 10 | 120 |
| bicarbonate HCO3− | 71.0 | 258 | 248 | 357 | n.a. |
Footnotes
* value for the area containing The Open University in Milton Keynes; - = too small to measure; n.a.= not availableActivity 1 Ions in drinking water
Question 1
This activity aims to access up-to-date information about ions in drinking water. You will access the web pages of the Drinking Water Inspectorate, DWI (2015) of England and Wales. (Hint: use the A-Z index.)
Search for information to answer the following questions:
Which ions in Table 1 are responsible for the hardness of water?
Answer
Cations such as calcium and magnesium.
Question 2
What is the World Health Organization (2015) guideline value for nitrate concentration in drinking water?
Answer
The World Health Organization (2015) guideline value for nitrate is 50 mg l−1 for drinking water and the EU also adopts this value.
Question 3
How does the concentration compare with the values for nitrate concentration in Table 1?
Answer
All the values for nitrate in Table 1 are well below 50 mg l−1.
Question 4
What is the allowed level of fluoride ions in drinking water? (Note this is often artificially added to water supplies.)
Answer
The maximum permitted value of fluoride in drinking water is 1.5 mg l−1.
You might wish to check the water quality report for your own water supplier. This is often available on the company website or see if the values for tap water in Milton Keynes in Table 1 have altered. Links to information on drinking water in other EU states can be found at European Commission (2015).
1.2 The toxicity of chemicals
As you have just considered several safe levels for chemicals in drinking water, it is appropriate now to discuss what is meant by toxicity. The German physician Paracelsus explored the relationship between dose and response. To paraphrase his conclusion: All things are poison and nothing without poison. Solely the dose determines that a thing is not a poison.
The toxicity of a chemical needs to be assessed in a way that relates dose to the receptor, such as an animal or human, and the time of exposure. Criteria, qualitative and quantitative statements of such relationships, are difficult to determine. For example, a high concentration may kill in a short time, but a lower concentration either may be lethal over a longer period than is measured, or may not kill but instead affect the behaviour or the susceptibility to environmental stress cumulatively over the lifetime of the receptor.
Figure 3 illustrates an approach to assessing toxicity, showing the connection between the dose given and the response to the chemical. This dose-response is measured by the percentage of rats killed by receiving a particular dose, expressed as milligrams per kilogram rat body weight. From this the LD50, the applied lethal dose responsible for killing 50% of the rat population, may be obtained (Table 2 shows some examples).
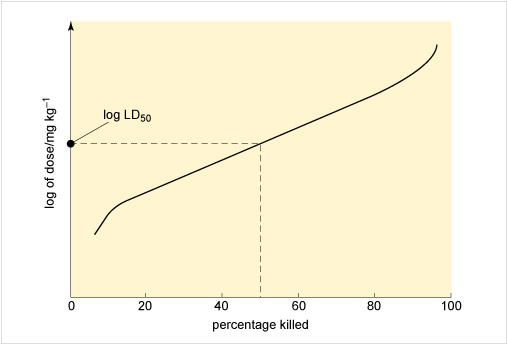
If the dose is expressed in terms of concentration, it is possible to record the percentage kill after different periods of exposure to varying levels of the chemical. Such a graph can yield the LC50, or median lethal concentration, which is the applied lethal concentration responsible for killing 50% of the rat population when administered as a single exposure.
In the case of the LD50, a distinction is made between short-term and long-term toxic action:
- An acute LD50 is measured after the administration of a single dose.
- A chronic LD50 refers to longer-term exposure following two or more doses given at different times.
- As LC50 values tend to be long-term measurements, they are more comparable to chronic rather than acute LD50 values.
- The effect varies depending upon how the chemical is administered, such as oral versus dermal administration.
A related value that is often quoted is the lethal dose low (LDLO) which is the lowest dose per unit of body weight (typically in mg kg−1) of a chemical known to have produced death in a particular receptor, such as a human or rat.
| LD50/mg kg−1 body weight | Examples | Classification |
|---|---|---|
| less than 25 | arsenic(III) oxide As2O3 | very toxic |
| sodium cyanide NaCN | ||
| thallium(I) sulfate Tl2SO4 | ||
| 25 to 200 | sodium nitrite NaNO2 | toxic |
| sodium fluoride NaF | ||
| 200 to 2000 | arsenic As | harmful |
| sodium nitrate NaNO3 | ||
| aluminium(III) fluoride AlF3 | ||
| indium(III) chloride InCl3 |
Compare: (a) the toxicity of As with As2O3 and (b) the various sodium salts, what does this suggest?
(a) The toxicity of As is lower compared to As2O3 which suggests that the oxidation number, often referred to as the oxidation state of an element, is important.
(b) The higher toxicity of NaCN compared to say NaNO2 suggests that the toxicity is determined by a contribution from each component ion present in an ionic compound, furthermore as this is oral toxicity these compounds will dissociate upon dissolution in the saliva or stomach acids. Indeed cyanide salts are generally very toxic compounds principally due to the cyanide anion.
For most kinds of toxicity, it is generally assumed that there is a dose below which no adverse effects will occur. For chemicals that give rise to known toxicological effects, a tolerable daily intake (TDI), over the whole lifetime without causing harm, can be derived as follows:
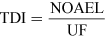
or
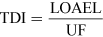
where UF is an uncertainty factor.
The NOAEL, no observable adverse effect level, defines the level of exposure to a chemical at which there is no biologically or statistically significant increase in the frequency or severity of any adverse effects in the exposed population compared with a control group. The closely related LOAEL, lowest observable adverse effect level, is the level where adverse effects are observed.
The guideline value (GV) is then calculated from the TDI as follows:

where bw = body weight, taken as 60 kg for adults, 10 kg for children and 5 kg for infants, p = fraction of the TDI allocated to drinking water, C = daily drinking water consumption (2 litres for adults, 1 litre for children, 0.75 litre for infants).
Toxicological information is usually found within the Materials Safety Data Sheet (MSDS) for commercial chemicals on the websites of chemical suppliers such as Fisher Scientific or Sigma Aldrich.
Problems have historically been reported from drinking well water polluted at 100 mg NO3− per litre. Assume the toxicity is due to the nitrate anion and the oral LDLO for sodium nitrate is 22.5 mg kg−1 for an infant, would a typical infant drink enough polluted water to be in any potential danger? Assume a 5 kg infant as suggested earlier and that an infant normally drinks 150 ml per kg per day.
Assuming 5 kg infant, so LDLO = 22.5 × 5 mg = 112.5 mg of NaNO3.
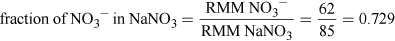 (Equation 5)
(Equation 5)So, NO3− LDLO = 112.5 × 0.729 mg = 82 mg
For polluted water containing NO3− at 100 mg l−1 to reach an LDLO of 82 mg requires drinking water of
 (Equation 6)
(Equation 6)to risk a potentially lethal acute dose.
Normally, an infant drinks 5 × 150 ml per day = 750 ml = 0.75 l.
Therefore, the infant would have been within 10% of a potentially harmful dose. However, Table 1 illustrates that the nitrate concentration is now not observed as high as 100 mg l−1 in the EU countries because the European Commission regulates the maximum amount of nitrate at 50 mg l−1.
Summary of Section 1
- Drinking water contains a mixture of anions and cations which are monitored and managed to avoid adverse health or environmental effects.
- The toxicity of a chemical to a receptor can be illustrated by a dose-response curve.
- The LD50 is the lethal dose responsible for killing 50% of the receptor population. The LC50 is the analogous lethal concentration.
- Toxicological effects observed in the short-term are termed acute while chronic effects arise from prolonged exposure.
- The lethal dose low (LDLO) is the lowest dose per unit of body weight of a chemical known to have produced death in a particular receptor.
- Tolerable daily intake or guideline values are often set for chemicals in drinking water.
2 Anions in water
There are several anions commonly present in water that require monitoring and management to avoid harm to human health and the environment as you saw in Table 1 and Activity 1. For example, fluoride is important in dental health as it forms a coating of the mineral fluorapatite on teeth which resists decay. However, if levels of fluoride are too high in drinking water then health problems may result. You will also study here nitrate, arsenate, and phosphate.
2.1 The nitrogen cycle
Cycling of nitrogen and its compounds through the environment involves a delicate balance of redox, atmospheric and biological processes (Figure 4) generally involving water as a solvent. Plants require nitrogen, which is absorbed in the form of nitrate or ammonium ions, for the synthesis of nitrogenous organic compounds, such as amino acids, which are incorporated into the tissues of the plant. However, some of the intrinsic nitrogen is lost upon removing a crop from the soil. This must be replaced to maintain the fertility of the soil.
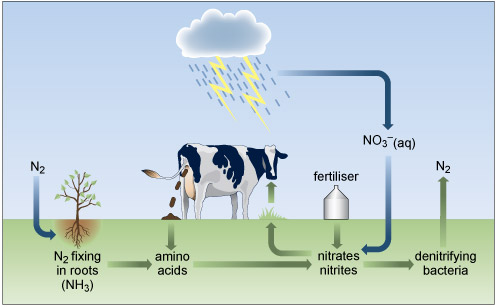
Nitrogen has a versatile redox chemistry and displays several oxidation numbers in Figure 4.
What are the oxidation numbers of the nitrogen in ammonia (NH3), nitrogen gas, nitrite (NO2−) and nitrate (NO3−)?
The oxidisation number of hydrogen is generally +1 and oxygen is generally −2. The oxidation numbers of nitrogen in these compounds aree −3, 0, 3 and +5, respectively, to maintain the charge seen on the compounds. Note nitrogen compounds display oxidation numbers ranging from −3 to +5.
Like other elements in the second row of the Periodic Table, the nitrogen atom can form double bonds both to other nitrogen atoms and to atoms of some other elements of the second row, such as boron, carbon and oxygen.
Determine the resonance forms in the Lewis structure of nitrite.
There are two resonance forms (Structure 1).
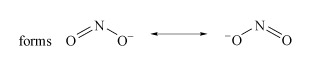 Structure 1
Structure 1
2.1.1 Natural nitrogen fixation
Despite a vast reservoir of nitrogen being readily available in the air, green plants cannot use it because of the high dissociation energy of the dinitrogen bond.
What is the bond order in dinitrogen, N2?
Three as dinitrogen contains a triple bond, N≡N.
Green plants depend on nitrogen fixation - the combination of atmospheric nitrogen with hydrogen and oxygen to form ammonium compounds or nitrates. On the roots of peas, beans and other members of the legume family, there are nodules (Figure 5), inside which live nitrogen-fixing bacteria of the rhizobia group. These bacteria fix about 108 tonnes of nitrogen per year worldwide, which is approximately 60% of all nitrogen fixed. This is a symbiotic relationship, with the bacteria providing the plant with nitrogen compounds, and the plant supplying nutrients to the bacteria.

To bring about cleavage of the nitrogen molecule requires a high energy input. Indeed, two molecules of the energy-transfer agent adenosine triphosphate (ATP; Section 3.3.2) are needed to bring about transfer of each electron. The reaction is catalysed by the enzyme nitrogenase, a complex molecule that incorporates molybdenum, iron and sulfur.
How many electrons are transferred when converting nitrogen to nitrate?
The oxidation number change is 0 (N2) to +5 (NO3−) suggesting the transfer of five electrons.
So how many molecules of ATP are needed to provide the energy for this conversion?
Each electron transfer requires two molecules of ATP, so ten ATP molecules in total.
Nitrogen compounds are produced in several other processes, the most obvious being for fertiliser, either farmyard manure or synthetic, such as ammonium nitrate (NH4NO3). Atmospheric lightning also causes some combination of oxygen and nitrogen (Equation 7) which forms nitrogen monoxide, which after further reactions leads to the passage of nitrogen into the soil as nitrates dissolved in rainwater.
The formation of gaseous oxides of nitrogen, NOx, from coal burning and internal combustion engines, is a process more commonly associated with detrimental environmental effects. So it should also be included.
The nitrogen cycle is completed by plant death, decay and bacterial denitrification, which returns nitrogen to the seas and the atmosphere. Originally fertilisers with a high nitrogen and phosphorus content originated from guano or bird excrement. For many years, naturally occurring sodium nitrate, NaNO3, from Chile was the main raw material for producing fertilisers.
2.1.2 Ammonia and synthetic nitrogen fixation
Ammonia is a colourless gas with a very strong characteristic smell. It was first produced industrially by the Haber-Bosch process in Germany (Equation 8).
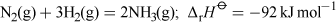
At room temperature the reaction is very slow, which means industrial processes must operate at high temperature (400-450 °C) and high pressure (8-35 MPa) and also require the presence of an iron catalyst. The fertilisers ammonium nitrate, NH4NO3, and ammonium sulfate, (NH4)2SO4, are manufactured by treating ammonia with either nitric or sulfuric acid, respectively.
Nitrogen is the first element of the second row of the Periodic Table to have a non-bonded pair of electrons in its normal valency state; this is typified by the ammonia molecule (Structure 2). Consequently, ammonia is a reasonably strong Lewis base (electron donor), particularly towards transition metal ions.
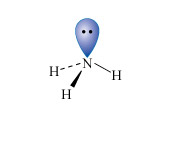
Nitrogen is also among the most electronegative elements and this affects both the physical properties and reactivities of its compounds.
Consider the boiling temperatures of the hydrides on descending Group 15: NH3, −33.4 °C; PH3, −87.7 °C; AsH3, −62.4 °C; SbH3, −18.4 °C. Why does ammonia have an uncharacteristically high value?
Ammonia has a higher than expected value due to the extra intermolecular forces or hydrogen bonds between its molecules.
Watch Video 1 and then answer the question following it.
Transcript: Video 1 The reaction of gaseous ammonia with hydrochloric acid.
After watching Video 1, suggest an equation for the reaction of ammonia with hydrochloric acid.
The white 'smoke' comprises small particles of ammonium chloride (Video 1):
NH3(g) + HCl(g) = NH4Cl(s)(Equation 9)
Ammonia forms an alkaline solution in water, often used in household cleaners.
Liquid ammonia is additionally a widely used non-aqueous solvent. Now watch Video 2 and answer the question following it.
Transcript: Video 2 The dissolution of sodium in ammonia.
What is seen upon the dissolution of sodium in ammonia in Video 2?
Alkali metals, such as sodium, dissolve reversibly to give blue solutions (in contrast to their more familiar vigorous reactions with water). The blue colour is thought to arise from electrons solvated by ammonia molecules.
2.1.3 Ammonium nitrate fertiliser
Nitric acid has many uses, but most is combined with ammonia to produce the important fertiliser ammonium nitrate. Nitric acid, HNO3, is made on a huge industrial scale by the Ostwald process: involving ammonia oxidation in two stages over a platinum metal catalyst, first to NO and then to NO2 (see Video 3):
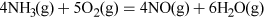
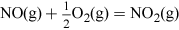
Transcript: Video 3 The oxidation of ammonia.
In Video 3 why did the platinum catalyst continue to glow?
The exothermic reaction in Equation 10 heated the platinum.
The NO2 is then dissolved in water to give a concentrated aqueous solution of the acid, and the NO produced in this step is recycled back into earlier stages. The overall reaction can be summarised by the following equation:
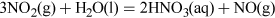
The anhydrous acid can be produced by distillation, and is a colourless pungent liquid.
Structure 3 shows the planar nitric acid molecule:
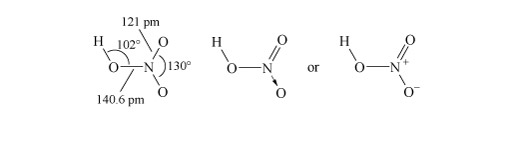
In dilute aqueous solution, nitric acid behaves as a typical strong acid, dissociating completely into H+ and NO3− ions.
Predict using valence-shell electron-pair repulsion, VSEPR theory (valence-shell electron-pair repulsion theory) t the shape of nitrate, NO3−.
The Lewis structure and structural formula of one resonance form of NO3− are in Structures 4 and 5. This adopts a planar shape because there are only three repulsion axes and no non-bonding pairs:
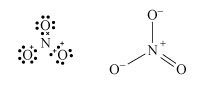 Structures 4 (left) and 5 (right)
Structures 4 (left) and 5 (right)
Ammonium nitrate fertiliser must be packed and handled with extreme care; in addition to being thermally unstable at high temperatures, its decomposition is catalysed by many inorganic and organic materials.
Strict regulations governing the storage and transportation of the chemical were introduced following explosions in 1947 aboard two American ships transporting fertiliser-grade ammonium nitrate to Europe. At the time of writing, in 2013, this remains the worst industrial accident in US history. In 2013 at Waco in Texas, a large explosion occurred at a fertiliser plant that stored ammonium nitrate.
Today, ammonium nitrate is used extensively in industrial explosives and propellants. For instance, commercial blasting explosive contains ammonium nitrate, fuel oil and TNT.
Video 4 shows that upon gentle heating ammonium nitrate decomposes.
Transcript: Video 4 Heating nitrates.
Taking it a stage further, we can demonstrate the oxidising power of potassium nitrate by melting some, and adding a piece of charcoal. In this vigorous reaction, the gases evolved are carbon monoxide, carbon dioxide, and nitrogen. This is not too far removed from the chemistry of gunpowder, where charcoal and sulfur act as fuels, which are oxidised highly exothermically by potassium nitrate.
Suggest an equation for the gentle heating of ammonium nitrate.
- NH4NO3(s) = N2O(g) + 2H2O(l)(Equation 14)
However, upon heating above about 250 °C, an alternative explosive decomposition occurs:
Potassium nitrate, known as saltpetre, is a component of gun powder (Video 4). The explosive material consists of sulfur and charcoal (a fuel), mixed with potassium nitrate (an oxidiser).
A simplified equation for the deflagration (a self-propagating burning surface reaction) of gunpowder is:
In Videos 3 and 4 you have seen examples of the production of some of the many oxides of nitrogen, namely nitrogen monoxide, NO, nitrogen dioxide, NO2, and dinitrogen monoxide, N2O. It is noteworthy that in biology NO adopts several key roles such as in the body where NO plays an important role in the regulation of blood pressure.
Nitrates of almost all of the metallic elements are known. They are all soluble in water which means that they are highly mobile in the environment. Consequently the EU regulates the application of nitrogen containing fertilisers through the EU Nitrates Directive.
2.1.4 Nitrate in natural water
Nitrate in water arises from excessive fertiliser use, as a waste product from many organisms and natural decay. The nitrate level in natural water is important as excess nitrate may cause problems from eutrophication. This is when the ecological balance of a body of water such as a lake is disturbed by excessive growth of algae at certain times of year. As the weather gets colder, the growth ceases, and the algae sink and decay, consuming oxygen dissolved in the water. This can result in an unpleasant smelling lake (largely due to the production of hydrogen sulfide) and, in an extreme case, the fish in the lake can die from oxygen starvation. Figure 6 shows algal growth in eutrophic water.

In drinking water the recommended maximum level for nitrate provides a safeguard against the disorder methaemoglobinaemia, which is potentially fatal for infants younger than six months. The toxicity occurs because the nitrate is reduced to nitrite (NO2−) which then interferes with the mechanism of transporting oxygen around the body by the protein haemoglobin. This reduction of nitrate is often associated with bacteria.
What is the change in the nitrogen oxidation number on going from nitrate to nitrite?
The nitrogen is reduced from an oxidation number of +5 to +3.
Excess nitrate can be removed in water treatment by ion-exchange chromatography (IEC, Figure 7) involving here water as the so-called mobile phase. IEC is based on a chromatography technique whereby dissolved species, or solutes, are adsorbed to varying extents onto the ionic sites of a so-called stationary phase which is an ion-exchange medium or resin (grey circle in Figure 7). One ionic species (the solute) is substituted for another from the stationary phase. If negatively charged ions are exchanged, it is called anion-exchange chromatography; the exchange of positively charged ions is called cation-exchange chromatography.
The stationary and mobile phases are chosen to selectively retain ionic species. The ionic strength of a solution is a function accounting for the concentration of all the different ions. By changing the ionic strength of the mobile phase, the ions in Figure 7 are eluted, or extracted into the mobile phase (here water), from the stationary phase. The ions bound by the stationary phase are eluted in order of the strength of binding, the most weakly bound ions being eluted first. The binding strength of the ions with the stationary phase depends on the differences in charge and charge density of the various ions present.
Here, nitrate ions are exchanged with chlorides on an ion-exchange resin (represented as R* in Equation 17 and as grey circles in Figure 7, see also Section 2.2.1):
Beads of the medium or ion-exchange resin contain a covalently bound group holding a positive charge, for example R−N(CH3)4+X− where R is a polymeric resin and X− is an anion which can be exchanged.
Why do you think the ion-exchange resin is regenerated by eluting with a concentrated sodium chloride solution? (See Figure 7)
With a high concentration of chloride ions the nitrate ions are exchanged and the initial chloride form of the ion-exchange resin is regenerated:
R*-NO3 + Cl−(aq) = R*-Cl + NO3−(aq)(Equation 18)
2.2 Arsenic pollution in the environment
Inorganic arsenic is naturally present at high levels in the groundwater of a number of countries, including Bangladesh, China, India, Mexico, and the USA. Note when articles discuss arsenic levels in natural waters they are talking about the oxoanions arsenite, AsO33−, and arsenate, AsO43−.
What are the oxidation numbers of arsenic in arsenite and arsenate?
Arsenate(III) and arsenate(V) or +3 and +5 respectively, which are bioavailable forms of arsenic.
The World Health Organization (2015) suggests a maximum safe level for drinking of arsenic of only 10 µg l−1.
Which vital element do you think arsenic might replace in the body?
Phosphorus displays similar chemistry as it is above arsenic in Group 15 and is indeed replaced by arsenic in organs and tissues within the body. For example, phosphorus can be substituted by arsenic in adenosine triphosphate (ATP) which plays a fundamental role in metabolism.
In natural waters one of the main sources of arsenic oxoanions is believed to be the weathering of pyrite, FeS2, minerals which contain a trace of arsenic. Weathering also oxidises some of the pyrite to form iron(III) hydroxide, Fe(OH)3, and sulfate. Remarkably, this iron(III) hydroxide then has a capacity to exchange its surface hydroxide anions and so is able to adsorb arsenite and arsenate anions from natural water effectively restricting their mobility in the environment. However, this adsorption on iron(III) hydroxide is pH-dependent, and so the arsenate ions can be re-mobilised into the environment by a pH change.
Which other anion in water do you predict iron(III) hydroxide might adsorb?
Phosphate, PO43−, is similar to arsenate as in both anions the central elements are in Group 15. So phosphate can be similarly absorbed.
2.2.1 Removal of arsenic oxoanions from drinking water
To remove arsenite and arsenate anions from water one method is to add particles of iron(III) hydroxide and allow time for the anion exchange to take place. However, where the water is pumped directly from a well for use, a better solution is to use a synthetic ion-exchange medium or resin as in Figure 8 and similar to what you previously saw for the removal of nitrate from water.
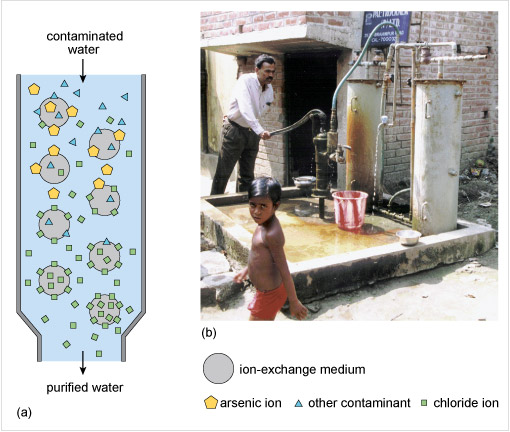
For arsenic contamination, the ion-exchange medium exchanges chloride ions, Cl−, for arsenate ions, AsO43−. Ion-exchange is almost instantaneous, a big advantage over adsorption processes using iron(III) hydroxide. After a few months the medium becomes saturated with arsenate ions and so is regenerated by eluting with a sodium chloride solution. This process presupposes a safe means of disposing of the eluted arsenate containing solution.
In practice, it has been observed that oxidation is beneficial before the removal of arsenic oxoanions. What is the chemistry behind this observation?
Oxidation favours formation of arsenic acid, H3AsO4, which is a stronger acid than arsenous acid, H3AsO3.
At neutral pH, which of the oxoanions, arsenite AsO33− and arsenate AsO43− will bind most strongly to a cation-exchange resin?
Arsenate, AsO43−, binds more strongly to a cation-exchange resin. Arsenate will be more fully deprotonated at neutral pH because arsenic acid is a stronger than arsenous acid. Consequently there will be a higher negative charge and greater electrostatic attraction to the cationic resin.
Summary of Section 2
- Nitrogen in the atmosphere is relatively inert due to its strong triple bond.
- Bacteria in the roots of plants fix nitrogen from the atmosphere.
- Industrially the Haber-Bosch process fixes nitrogen:
N2(g) + 3H2(g) = 2NH3(g)(Equation 8)
- Nitric acid is produced by the Ostwald process:
(Equation 11)(Equation 12)(Equation 13)
Nitric acid is then used to make ammonium nitrate fertiliser.
- Excess levels of nitrate, NO3−, in water can result in eutrophication in the environment. While high levels of nitrate in drinking water are dangerous for infants because the nitrate can be reduced to nitrite in the body which may interfere with oxygen binding by haemoglobin.
- Arsenic levels in natural waters correspond to the oxoanions arsenite, AsO33-, and arsenate, AsO43-.
- Excess nitrate or arsenate can be removed from water by ion-exchange chromatography.
- Fluoride is important in dental health but if levels are too high in drinking water then health problems may result.
3 Phosphorus compounds in water
Phosphorus compounds are hugely important in nature, for example deoxyribonucleic acid (DNA) is a polymer of alternating phosphate and deoxyribose groups. In this section you will see that phosphorus forms firstly relatively simple anions such as phosphate, PO43-. Then you will explore the chemistry of polyphosphates such as adenosine triphosphate (ATP) that was mentioned in Section 2.1.1.
3.1 Phosphates in the environment
Most detergents or surfactants, comprise long molecules with hydrophobic (water-repelling) and hydrophilic (water-attracting) parts as indicated in the example in Structure 6. The basic action is for the organic hydrophobic section to bury itself in the dirt, and the hydrophilic section then allows the insoluble dirt to 'dissolve' in water.
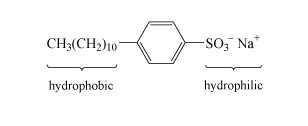
Detergents are used in combination with so-called builders, which soften hard water. Hard water contains significant concentrations of calcium and magnesium salts, which replace the sodium ions in the detergent molecule. The dipositive metal ions cause the long detergent molecules to clump together and precipitate out as a scum.
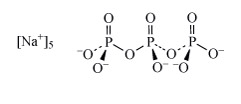
Sodium tripolyphosphate (Structure 7 see also Section 3.2.1), was used in detergents to complex magnesium and calcium ions and hence remove them from solution. Due to environmental concerns over such polyphosphates acting as nutrients for algae (leading to eutrophication), detergent manufacturers have phased out the use of so-called phosphate builders. Substitute detergent builders include zeolites, such as ZSM-5 NanAlnSi96−nO192·16H2O (0 n

3.1.1 Phosphoric acid and fertilisers
Phosphoric acid is manufactured from phosphate minerals, and the pure acid forms low-melting crystals (Tm 42 °C). Commercial phosphoric acid is 85% phosphoric acid in water; this forms a syrup because the acid molecules are hydrogen-bonded to water molecules.
Most phosphoric acid is used to manufacture fertiliser. For example, so-called triple superphosphate fertilisers are manufactured from calcium phosphate-containing rock, such as Ca3(PO4)2, and phosphoric acid:
The farming of crops depletes soils of essential nutrients, such as phosphate and nitrate, which are replenished by applying fertilisers containing suitable inorganic compounds. Consequently, eutrophication from excess phosphate and nitrate in rivers and lakes remains an issue but now often occurs due to water run-off from agricultural land.
Figure 10 shows the excessive algal growth in the Mediterranean arising from excess nutrients in the water. These nutrients are regulated under the EU Water Framework Directive. For example, agricultural practices are altered to minimise pollution from applying nutrients to soils.

Phosphate can be removed from water by precipitation with lime, Ca(OH)2, forming hydroxyapatite (Equation 20), the same material which comprises bone and teeth:
Phosphate is recovered from wastewater during chemical sewage treatment, often as struvite, NH4MgPO4.6H2O, which can then be used as a fertiliser. Phosphate can also be recovered during biological water treatment where it is used in the growth of cell membranes, a process which ultimately forms biological solids or so-called sludge which can be used as a fertiliser.
3.2 Oxoacids
Many p-block elements form oxoacids but here the focus will be on those of phosphorus with a few illustrative examples of other elements.The chemistry of phosphorous compounds in natural water and the body is not quite as simple as it has been treated so far, so the topic will be expanded upon here.
For instance, there are several oxoacids of phosphorus. The three most important are phosphoric acid, H3PO4, phosphorous acid, H3PO3, and hypophosphorous acid, H3PO2. Table 3 lists other names for these acids.
What is the oxidation number of phosphorus in H3PO4, H3PO3, and H3PO2?
The oxidation number is +5, +3 and +1, respectively.
Therefore H3PO4, H3PO3, and H3PO2 are also called phosphoric(V), phosphoric(III) and phosphoric(I) acid respectively.
| Formula and common name | Common anion name | Structure |
|---|---|---|
H3PO4 phosphoric acid or orthophosphoric acid | phosphate or orthophosphate | 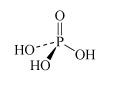 |
H3PO3 phosphorous acid or phosphonic acid | phosphite or phosphonate | 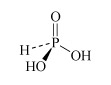 |
H3PO2 hydrophosphorous acid or phosphinic acid | hypophosphite or phosphinate | 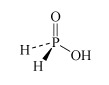 |
H4P2O7 diphosphoric acid | diphosphate | 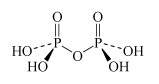 |
(HPO3)n (in the limit where n = ∞) metaphosphoric acid | metaphosphate |  |
Some acids are polyprotic (or taking the alternative viewpoint polybasic) meaning they ionise stepwise, unlike hydrochloric acid, HCl, which is a monoprotic acid. Note only the protons attached to oxygen are ionisable. Phosphoric acid is triprotic undergoing three successive ionisations:
As phosphoric acid is triprotic, it can form three series of salts with a metal such as sodium: the dihydrogen phosphate, the hydrogen phosphate and the normal phosphate.
What would you expect to be the sodium salts of phosphorous acid, H3PO3?
Phosphorous acid is diprotic, forming the phosphite salts NaH2PO3 and Na2HPO3. Note one of the hydrogen atoms in H3PO3 is directly attached to the phosphorus and consequently does not undergo ionisation.
Note that Equations 21-23 have the form:
The pairs acid(1)/base(1) and acid(2)/base(2) are called a conjugate acid and base. Thus, H3O+ and H2O are a conjugate acid and base pair; so are H3PO4(aq) and H2PO4−(aq). An acid is transformed to its conjugate base by losing a proton and vice versa.
3.2.1 Polyacids
A large variety of phosphorus acids are derived from 'polyacids', which contain two or more acidic phosphorus centres (see Section 3.3).
Oxoacids by definition contain a covalent AO-H bond, which can dissociate to give a proton and an oxoanion:
There may also be one or more terminal oxygen atoms, so the general formula of oxoacids is A(O)t(OH)n, where t can equal 0. By this formulation, sulfuric acid, H2SO4, is written as S(O)2(OH)2 (where t = 2, n = 2), and boric acid as B(OH)3 (where t = 0, n = 3).
What are the values of t and n for phosphoric acid?
From Table 3, the formula of phosphoric acid can be rewritten as P(O)(OH)3, giving t = 1 and n = 3.
These covalent hydroxo compounds have available a wide range of structural possibilities, which is the reason for the existence of a relatively large number of oxoacids. The variables are as follows:
- There may be several -OH groups in the acid, each one of which can dissociate to form a proton and an oxoanion. For example, in the three successive ionisations of phosphoric acid, H3PO4 (Equations 21-23): each of the species on the left-hand side of the three equations is a different oxoacid, and each of the equilibrium has a different dissociation constant.
- The equilibrium constant for dissociation of the first proton (stage 1), or the 'first dissociation constant', is given the symbol K1 (7.5 × 10−3 mol l−1). The second dissociation constant is K2 (6.2 × 10−8 mol l−1). The third dissociation constant is given the symbol K3 (1.0 × 10−12 mol l−1).
- Oxoacids may undergo condensation reactions to form dimers, trimers and polymers. The term 'condensation' refers to the reaction of two or more molecules to form a larger molecule, with the elimination of a small molecule, most often water. Thus, phosphoric acid, H3PO4, can self-condense to produce diphosphoric acid and triphosphoric acid:
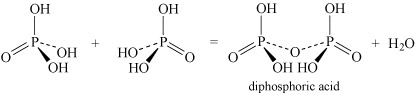 Equation 26
Equation 26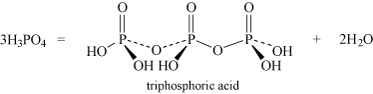 Equation 27
Equation 27It can also form higher polymers described by the general term 'metaphosphoric acid'.
- The central element, A, may exist in more than one oxidation number. Remember, oxygen stabilises high oxidation numbers. In fact, the range of oxidation numbers found in compounds with oxygen is wider than that with any other element except fluorine. Phosphorus, for example, forms oxoacids in oxidation numbers +5, +3 and +1.
- A final complication, arising with sulfur, is that this Group 16 element can take the place of oxygen in an oxoacid, so that there is the possibility of one or more S-S bonds, in what is then called a thioacid.
3.2.2 Nomenclature of oxoacids
Traditionally oxoacid formulas are written with hydrogen first, which conceals the fact that hydrogen is often bonded to oxygen. The rarely used formula (OH)3PO is a more meaningful formulation of phosphoric acid than H3PO4. The nomenclature is further complicated by the fact that the inorganic acids and their organic derivatives also have different common names; for instance, phosphorous acid in inorganic chemistry becomes phosphonic acid for its organic derivatives. To help you through this topic the prefixes and suffixes are in bold italics and the most common name is always used.
Historically, where the central element forms oxoacids in two oxidation numbers:
- the higher state is indicated by the suffix -ic
- the lower state is indicated by the suffix -ous.
For example phosphoric acid, H3PO4, and phosphorous acid, H3PO3, have phosphorus oxidation numbers +5 and +3 respectively.
If more than two oxidation numbers are involved, the prefixes per- and hypo- are used as well:
- per- denotes the highest oxidation number
- hypo- denotes the lowest oxidation number.
For example the oxoacids of chlorine are shown in Table 4.
| Formula | Oxidation number | Name |
|---|---|---|
| HClO4 | +7 | perchloric |
| HClO3 | +5 | chloric |
| HClO2 | +3 | chlorous |
| HClO | +1 | hypochlorous |
Oxoanions derived from -ic acids are given the ending -ate and from -ous acids are given the ending -ite.
To practise, name the following anions OCl−, ClO4− and NO2−.
The ions are hypochlorite, perchlorate and nitrite.
Condensed forms of oxoacids are also distinguished by prefixes:
- ortho- refers to the 'monomeric', or most highly hydroxylated, form
- meta- refers to the 'polymeric', or least highly hydroxylated, form
- di- and tri- in this context logically refer to 'dimers' or 'trimers'.
The oxoanions follow this labelling convention, for example orthophosphates, PO43−, diphosphates, P2O74−, and triphosphates, P3O105−. Additionally, the prefix cyclo- or catena- distinguish cyclic from linear condensed anions, respectively.
3.2.3 Prediction of formulas
For an element in its highest oxidation number (and sometimes others), it is possible to predict the formula of its orthoacid from its coordination number considerations. The condensed oxoacid formulas are then easily derived by subtracting the appropriate number of water molecules.
Third and fourth-row elements prefer four-coordination in their oxoanions. For example, using the oxidation number approach, you can see that when phosphorus(V) coordinates four O2− ions to form a complex ion of stoichiometry PO4, the resultant charge on this oxoanion is (+5 − 8) = −3. Thus, the formula of the corresponding neutral oxoacid is H3PO4.
Predict the formula of the oxoacid formed by sulfur(VI).
Sulfur(VI) coordinates four O2− ions to form a complex ion of stoichiometry SO4, with the resultant charge (+6 − 8) = −2, so the oxoacid is H2SO4.
The chlorine(VII) oxoanion, with stoichiometry ClO4, has the charge (+7 − 8) = −1, so only one proton is required for the formation of the neutral oxoacid HClO4.
For oxoanions of the second Period three-coordination is the norm, as it allows for the formation of a planar assembly with extensive π bonding, e.g. carbonate, CO32−.
The above arguments often fail to predict the formulas of oxoacids of elements in lower oxidation numbers, because the preferred coordination number is either not achieved, or is achieved only by formation of a direct link between the central atom and hydrogen.
3.3 Condensation of oxoacids
The tendency for an oxoacid to polymerise by condensation is most marked in the less acidic (more highly hydroxylated) acids. Consequently there are many stable condensed forms of silicic and boric acid which are found in minerals.
Condensation is most marked in structures where the charge on the uncondensed anion is high, because it is able to reduce the charge density on the anion. For example SiO44− dimerises as follows:
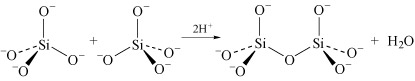
In the monomer there are four negative charges for four oxygen atoms; in the dimer there are six negative charges and seven oxygen atoms; in the trimer (Structure 8) there are eight negative charges and ten oxygen atoms.
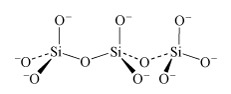
In the limiting case of the infinite polymer, there are two negative charges to every three oxygen atoms.
What is the repeat unit in Structure 8 upon double protonation of the individual unit?
In the case of protonation by two H+ per Si, the repeat unit is [H2SiO3].
This is the structural unit found in the pyroxene minerals (Figure 11). Here shared SiO4 tetrahedra can be assembled into chains, double chains, sheets, rings and three-dimensional networks to give an amazing variety of structures for crystalline silicate minerals.

3.3.1 Polyphosphates
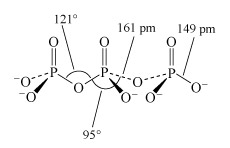
Phosphate tetrahedra, PO43−, can link up by oxygen-sharing to give polyphosphates in the form of rings and chains (Figures 12 and 13). The phosphates, unlike the silicates, contain a central atom with a maximum valency of five. Thus, the maximum number of oxygen atoms that each tetrahedron can share is three, since there must always be one vertex of the tetrahedron that is a terminal oxygen, bound as P=O.
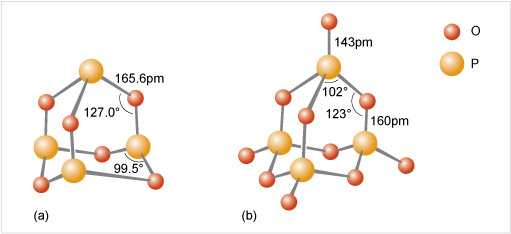
Consider phosphoric oxide, P4O10 (Figure 12b), how many P-O-P linkages does each phosphorus exhibit?
Each phosphorus atom forms the maximum three P-O-P linkages.
Condensed phosphates can be formed if phosphoric oxide is treated with limited amounts of water; or by dehydrating phosphorus oxoacids or their salts by heating.
Two molecules of phosphoric acid can condense by loss of a water molecule; the two tetrahedra share one oxygen, giving an acid of formula H4P2O7, known as 'pyrophosphoric', or (more correctly) diphosphoric acid. In practice, this can be obtained by heating phosphoric acid ('pyro' comes from the Greek word for fire). Continuation of this process gives triphosphoric acid, H5P3O10, and, eventually, chain polymers called metaphosphoric acids, which contain repeating [HPO3] units. In Figure 13 the ring polymers which are also shown are also called metaphosphoric acids.
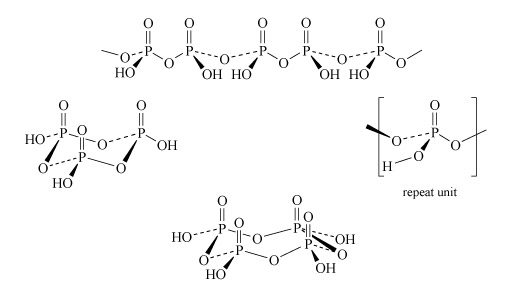
The P-O-P link in polyphosphates is readily hydrolysed; in excess water, metaphosphates revert to orthophosphate. So, unlike the condensed silicates, polyphosphates are never found as minerals. The hydrolysis of polyphosphates is an important source of energy within the body (Section 3.3.2).
Sodium polyphosphates are the best known; the general reaction for their formation is the dehydration of sodium dihydrogen phosphate by heating. The temperature of dehydration controls the nature of the product (Figure 14).
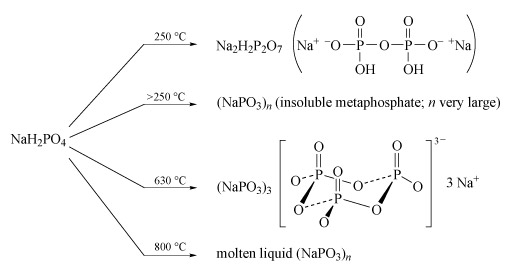
- Partial dehydration at low temperature (c. 250 °C) produces disodium dihydrogen diphosphate, Na2H2P2O7, a substance used in baking powders as a slow-acting acid for the controlled release of CO2 gas, which produces the aerated texture of cakes.
- Long-chain polyphosphates are produced by heating NaH2PO4 above 250 °C.
- The sodium salt Na3P3O9, containing the cyclic P3O93− anion, is formed when NaH2PO4 is heated to 600-640 °C and the melt then maintained at 500 °C.
- High-temperature dehydration (800 °C) above the melting temperature of NaH2PO4 (628 °C) yields a liquid with very high relative molecular mass, which gives slightly soluble solids of formula (NaPO3)n:
nNaH2PO4 = (NaPO3)n + nH2O(Equation 29)
- Heating together a 2 : 1 mixture of Na2HPO4 and NaH2PO4 at 450 °C produces a chain of only three phosphate units:2Na2HPO4 + NaH2PO4 = Na5P3O10 + 2H2O(Equation 30)
This triphosphate ion, P3O105−, is illustrated in Structure 9.
3.3.2 Adenosine triphosphate
After deoxyribonucleic acid, DNA (which is a phosphodiester), adenosine triphosphate (ATP, Structure 10), is probably the most important phosphorus-containing molecule in the human body.
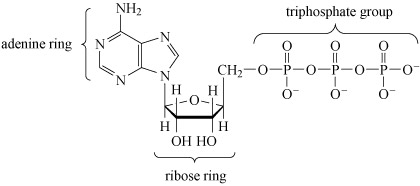
ATP is used to drive many biochemical reactions and cellular processes that require the input of energy; these include cell division and muscle contraction.
Energy is obtained from ATP when it is hydrolysed to adenosine diphosphate (ADP) and free phosphate ions. Equation 31 is thermodynamically favourable, having a free energy change,
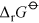 , of about −140 kJ mol−1.
, of about −140 kJ mol−1.
Note that when bonded together the adenine and ribose rings are referred to as adenosine.
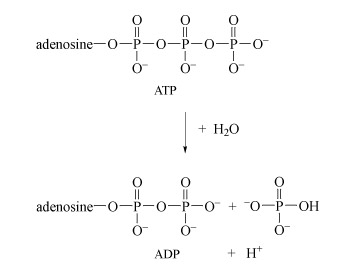
ATP is synthesised by the reverse process - the addition of phosphate to ADP. The energy input for this reaction comes from the breakdown of organic fuel molecules, such as glucose.
The negative charge on ATP, ADP and DNA is counterbalanced by cations, usually Mg2+. Hence, both ATP and DNA may be regarded as magnesium complexes.
Summary of Section 3
- Phosphate is used in fertilisers and excess phosphate in natural water can cause eutrophication.
- Phosphorus has several acids including phosphoric and phosphorus acid. Note the higher oxidation number, or oxidation state, is indicated by the suffix -ic and the lower oxidation state by the suffix -ous.
- Generally for oxoacids if more than two oxidation numbers are involved, the prefixes per- and hypo- are used as well where per- denotes the highest oxidation number and hypo- the lowest oxidation number.
- Some acids of phosphorus are polyprotic. For instance, phosphoric acid:H3PO4(aq) = H+(aq) + H2PO4−(aq)(Equation 21)H2PO4−(aq) = H+(aq) + HPO42−(aq)(Equation 22)HPO42−(aq) = H+(aq) + PO43−(aq)(Equation 23)
- Deprotonation of an acid yields its conjugate base or anion. In Equation 21 H2PO4− is the conjugate base of phosphoric acid.
- Oxoacids and oxoanions can polymerise by condensation.
- Phosphorus also forms polyacids containing two or more acidic phosphorus centres.
- Polyphosphate can form chain or ring polymers.
- Adenosine triphosphate (ATP) stores energy in the body which is released upon its hydrolysis to adenosine diphosphate (ADP).
- The negative charges on ATP, ADP and DNA are counterbalanced by cations, usually magnesium.
4 Cations in water
Sodium, potassium, magnesium and calcium are found in natural water (Table 1). The most important sources of calcium are the mineral deposits of calcium carbonate, CaCO3, which are formed from the fossilised remains of long-dead marine organisms. Examples include the minerals limestone and chalk.
Dissolved carbon dioxide makes rainwater slightly acid.
Suggest an equation for the dissolution of carbon dioxide in water.
- CO2(g) + H2O(l) = H2CO3(aq)(Equation 32)
A carbonic acid solution is weakly acidic:
H2CO3(aq) = H+(aq) + HCO3−(aq)(Equation 33)Consequently, the rock limestone is very slightly soluble in rain:
CaCO3(s) + H+(aq) = Ca2+(aq) + HCO3−(aq)(Equation 34)
This accounts for the levels of both calcium and bicarbonate (HCO3−) ions seen in natural water samples (Table 1). Furthermore, this dissolution of calcium carbonate contributes to the hardness of water.
Water hardness is often due to the presence of dissolved calcium and magnesium salts. For instance, calcium hydrogen carbonate upon heating is converted to calcium carbonate. This calcium carbonate is insoluble and deposits in appliances, such as kettles.
Human derived acid rain can weather minerals by, for instance, converting calcium carbonate (Figure 15) into calcium sulfate, CaSO4, leading to harder water. The resulting volume change leads to surface cracking and new conduits for water to percolate. Freezing of this water leads to further damage. Such acid rain can arise from the combustion of sulfur-containing fuels because this yields sulfur dioxide, SO2, which upon oxidation can yield sulfuric acid. Consequently sulfur compounds are removed from fuels before combustion where possible, often using zeolites.

Suggest an equation for the reaction of calcium carbonate with sulfuric acid.
- CaCO3(s) + H2SO4(aq) = Ca2+(aq) + SO42−(aq) + H2O(l) + CO2(g)(Equation 35)
4.1 Aqueous chemistry of aluminium
Aluminium is a metal forming an aqueous cation, unlike the non-metal boron found above it in Group 13. For example, hydrates of aluminium sulfate, Al2(SO4)3, can be made by dissolving bauxite, Al2O3.3H2O, in sulfuric acid and evaporating the solution. Dissolving such sulfates in water forms Al3+(aq) (Figure 16):
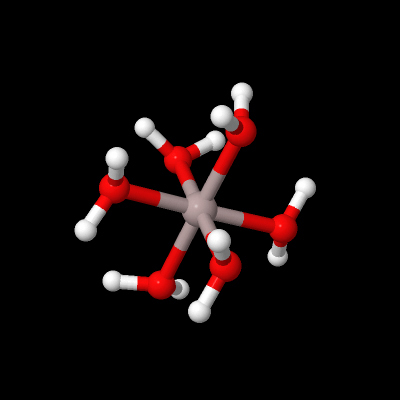
What is the coordination geometry of aluminium in Figure 16?
Al3+(aq) is shorthand for the octahedral coordination complex Al(H2O)63+.
Careful addition of aqueous sodium hydroxide to this solution will first precipitate insoluble aluminium hydroxide:
The oxide is produced by filtering and heating the hydroxide:
Both oxide and hydroxide are unusual in being amphoteric. Hence they will first dissolve in and neutralise acids:
Watch Video 5 to determine how else an amphoteric hydroxide (or oxide) behaves.
Download this video clip.Video player: Video 5Transcript: Video 5 The reaction of aluminium(III) ions with hydroxide ions.
End transcript: Video 5 The reaction of aluminium(III) ions with hydroxide ions.NARRATORThis is an aqueous solution of potassium aluminium sulfate, often referred to as 'alum'. It contains the aluminium three plus ion. If we add dilute sodium hydroxide, we precipitate aluminium hydroxide, an insoluble base used in indigestion tablets. However, in excess alkali, we form the soluble aluminate ion.Video 5 The reaction of aluminium(III) ions with hydroxide ions.Interactive feature not available in single page view (see it in standard view).It will dissolve in an alkali and will neutralise it.
As excess sodium hydroxide is added to the precipitate that is initially formed in Equation 37, the precipitate dissolves to form the tetrahydroxyaluminate ion:
This shows a close resemblance of Al(OH)3 to Be(OH)2, which is also amphoteric. Note that the bases ammonia and sodium carbonate are not strong enough to cause this dissolution.
4.1.1 Aluminium sulfate in water treatment
The Group 1 and Group 2 metals form carbonates, with those of Group 2 being insoluble in water. Aluminium carbonate, Al2(CO3)3, however, cannot be prepared; if aluminium sulfate is added to a solution containing carbonate or hydrogen carbonate ions, Al(OH)3 is precipitated and carbon dioxide is evolved:
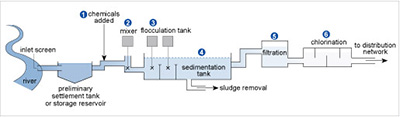
Figure 17 shows the stages in a typical water treatment process for river water. In stages 1 and 2 chemicals are added and mixed. Then in stage 3 flocculation or aggregation occurs involving large amounts of aluminium sulfate to clear the water of fine suspensions that are difficult to filter off, such as clay particles. The tiny particles usually carry surface negative charges, which repel each other and so remain in suspension.
The positively charged aluminium ions get between the negative particles, counteracting the repulsion and encouraging flocculation. Then, when Reaction 42 occurs because of the HCO3− ions usually present in natural waters, the particles sediment with the precipitate of aluminium hydroxide that forms in the sedimentation tank in stage 4. The water is filtered in stage 5 before disinfection in stage 6 via chlorination.
What role is Al3+(aq) playing in Equations 41 and 42?
Acids liberate CO2 from carbonate and hydrogen carbonate solutions, so here Al3+(aq) acts as an acid.
Then, Equation 43 becomes:
The positively charged alumnium ions get between the negative particles, counteracting the repulsion and encouraging flocculation. Then, when Reaction 42 occurs because of the HCO3 ions usually present in natural waters, the particles sediment with the precipitate of aluminium hydroxide that forms in the sedimentation tank in stage 4. The water is filtered in stage 5 before disinfection in stage 6 via chlorination.
Natural water generally requires disinfection before drinking to kill pathogenic or disease-causing microorganisms. Ultraviolet irradiation is one disinfection method and the major chemical disinfectants are ozone and chlorine. These compounds are thought to kill microorganisms by rupturing the cell membrane and reacting with proteins and enzymes within the cells. Once the chemical structures of proteins and enzymes have been altered they may either fall apart or adopt an unnatural state. Consequently, they fail to perform their roles and so the cell or bacteria dies.
Why can [Al(H2O)6]3+ be described as a Brønsted-Lowry acid in Equation 43?
Three of the six water molecules that were attached to the aluminium have been lost, but the other three have acted as proton donors, leaving aluminium associated with hydroxide ions instead of water molecules.
A more obvious sign of the acidic character of [Al(H2O)6]3+ is the fact that aqueous solutions of aluminium sulfate are acidic, unlike those of, say, sodium sulfate:
Again, a water molecule coordinated to Al3+ is transformed into an OH− ligand; at the same time, hydrated protons, H3O+, are generated.
4.1.2 Aluminium and biology
Aluminium minerals such as bauxite are biologically unavailable due to their insolubility in water. In the course of evolution, this would inevitably have limited the bioavailability of aluminium to living organisms. Aluminium is consequently not an essential element for humans in their normal metabolism. However small amounts of aluminium are found in most people's diet, let us now consider how this might arise?
Watch Video 6. Determine how aluminium reacts with both acid and alkali.
Download this video clip.Video player: Video 6Transcript: Video 6 Reaction of aluminium with acid and alkali.
End transcript: Video 6 Reaction of aluminium with acid and alkali.NARRATORAluminum is an amphoteric metal. It reacts with both acids and alkalis. On the left is an aqueous solution of sodium hydroxide. And on the right is dilute hydrochloric acid. Both reactions produce hydrogen.Video 6 Reaction of aluminium with acid and alkali.Interactive feature not available in single page view (see it in standard view).Aluminum is soluble in both acids and alkalis, dissolving in both hydrochloric acid and sodium hydroxide, liberating hydrogen and finally giving clear, colourless solutions:
2Al(s) + 6H+(aq) = 2Al3+(aq) + 3H2(g)(Equation 45)2Al(s) + 6H2O(l) + 2OH−(aq) = [Al(OH)4]−(aq) + 3H2(g)(Equation 46)
Aluminium metal forms a protective oxide film upon exposure to the air and so does not react further. Therefore, as aluminium is fairly inert, it is commonly used in cookware, especially as it is an excellent conductor of heat.
How might aluminium in cookware be solubilised? Also, consider if aluminium(III) is a hard or a soft cation.
Aluminium can be solubilised from cookware by heating acidic solutions, such as those containing citric acid, Structure 11). Aluminium(III) is a hard cation, similar to the iron(III) ion. Consequently it is bound and solubilised by hard chelating ligands in food such as citric acid.
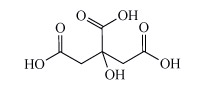 Structure 11
Structure 11
Aluminium has been associated with several neurodegenerative diseases although its role remains controversial. The World Health Organization (2015) sets a tolerable daily intake of aluminium for a 60 kg adult at 60 mg. For most people, the mass actually ingested daily is about 10 mg. This aluminium is mostly excreted in the faeces and is not taken up by the body. That which passes across the gastrointestinal barrier into the blood stream is dealt with by the kidneys (Figure 18). However, there is a small accumulation in the whole body, including the brain and lungs.
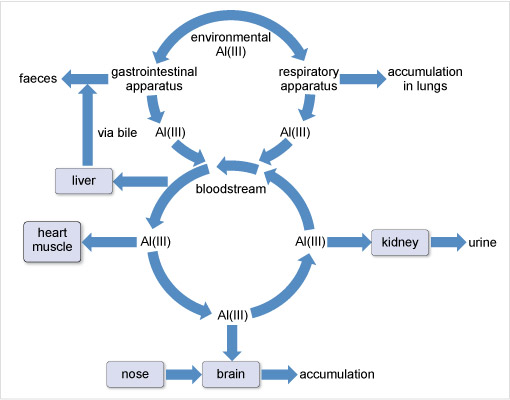
There is no doubt that aluminium can damage people with impaired kidney function. The condition called dialysis dementia was first noticed in patients who had received long-term haemodialysis for renal failure. Its symptoms included speech disorders, memory loss, convulsions and seizures, followed, in some cases, by death within a year. The incidence of the disease was highest when the municipal water used in the dialysis contained high concentrations of aluminium. Aluminium is therefore considered a potential neurotoxin.
4.1.3 Aluminium toxicity
The exact mechanism of aluminium toxicity remains controversial. Aluminium has been shown to disrupt lipid membrane fluidity, perturb iron, magnesium, and calcium regulation or so-called homeostasis, and cause oxidative stress by upsetting the normal treatment of unwanted oxidising radicals by the body.
Why is the observation that aluminium causes oxidative stress perhaps surprising?
Alumunium, unlike transition metals, does not have redox chemistry as it only forms the +3 ion. Therefore it is thought that aluminium(III) ions interfere with enzymes which counter reactive oxygen species (ROS) in the body.
Glycolysis is a nearly universal pathway in biological systems which aluminium can influence. Glycolysis includes a key step in which adenosine triphosphate (ATP) loses its terminal phosphate group to a glucose molecule and becomes adenosine diphosphate (ADP) (Figure 19).
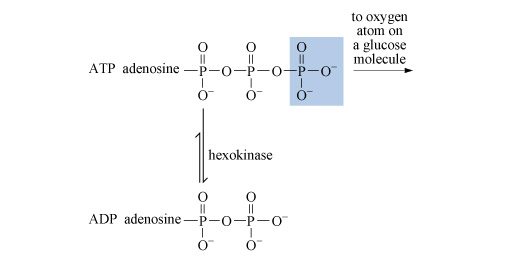
The process is catalysed by an enzyme called hexokinase. Before the ATP can be accepted by the enzyme, it must become complexed to a magnesium ion, Mg2+, through oxygen atoms attached to phosphorus atoms 2 and 3 (Structure 12). The exact way in which the magnesium ion bridges the two terminal phosphates is uncertain, but in this complexed state, the ATP is accepted by the enzyme. The magnesium ion then moves from the bridging position between phosphate groups 2 and 3 to one between groups 1 and 2. This exposes the terminal phosphate group to removal, and transfer to glucose.
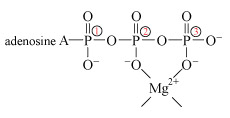
The change of bridging position is possible because Mg2+ does not bind particularly strongly to the oxygen donor sites. Now suppose substantial amounts of dissolved aluminium are present:
What ion is then available for binding to the oxygen donors?
Al3+(aq), which from simple electrostatic arguments will bind more strongly than Mg2+ to oxygen donor ligands.
Consequently Al3+ will bridge phosphate groups 2 and 3 like Mg2+, but because of the stronger binding, it is reluctant to shift to bridge groups 1 and 2 upon binding by the enzyme. Loss of the terminal phosphate is therefore impaired.
Whether this explanation of aluminium neurotoxicity is the whole story or not, it still points to a useful general idea about poisons. Their key feature is a resemblance to some chemical species (in this case Mg2+) that is essential to a biological process, and which is similar enough to gain access to a metabolic process. The poison once incorporated produces differences from the essential species and the metabolic process is blocked.
In natural waters the concentration of aluminium is increased by acid rain. When the hydrogen ions of acid rain fall upon soils that are naturally quite acidic, they replace other positive ions bound into the soil structure, such as Al3+.
For instance, the common mineral feldspar occurs widely in many different rock types, and is broken down during weathering by both physical and chemical degradation processes. Feldspar typically weathers as follows:
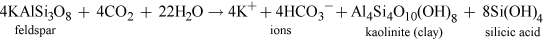
Kaolinite can react with water to give aluminium hydroxide (gibbsite) and hydrated silicic acid. The aluminium hydroxide dissolves in the presence of hydrogen ions.
Suggest an equation for the dissolution of aluminium hydroxide in acid.
From earlier:
Al(OH)3(s) + 3H+(aq) = Al3+(aq) + 3H2O(l)(Equation 48)
This aluminium contamination is a major contributor to the decline of fish stocks in lakes. The Al3+ ion appears to bind to oxygen-donor ligands, such as the phosphate groups in phospholipids, at the surface of fish gills. This makes the membrane of gill cells more permeable to further Al3+, which can then enter the cells and replace Ca2+ ions on key proteins. The regulation of the cell concentrations of other ions, such as Na+, is also disrupted. At the same time, the increased concentration of aluminium in gill cells leads to precipitation of aluminium hydroxide, mucus formation and breathing difficulties.
4.1.4 Toxicity of thallium(I) compounds
Another Group 13 element thallium, and especially thallium(I), is extremely poisonous. Thallium poisoning has been linked to industrial emissions from the burning of coal and the smelting of thallium-rich sulfide ores. Consequently the United States Environmental Protection Agency sets the safe level of thallium in drinking water at 0.5 ppb. When ingested, thallium seems to follow potassium in its metabolism, and it probably interferes with vital roles played by potassium in the nervous system. Note the ionic radius of Tl+ (160 pm) resembles that of K+ (152 pm). Again, this supports the general explanation of toxicity: the nervous system mistakenly accepts thallium(I) instead of potassium ions. Essential biological processes are then blocked because thallium(I) is an inadequate substitute.
How similar is the chemistry of thallium(I) and potassium ions shown in Video 7?
Download this video clip.Video player: Video 7Transcript: Video 7 A comparison of some reactions of thallium(I) and potassium ions.
End transcript: Video 7 A comparison of some reactions of thallium(I) and potassium ions.NARRATORFirst, aqueous sodium hydroxide is added to a solution of potassium sulfate. And then to a solution of thallium(I) sulfate. Now, an aqueous carbonate solution is added to potassium sulfate. And then to thallium sulfate. The hydroxide and carbonate of thallium(I) are soluble, like those of potassium. This is unusual.Soluble metal hydroxides and carbonates are rare. Potassium also forms an insoluble tetraphenylborate. Will thallium resemble it in this respect? The tetraphenylborate is added first to potassium sulfate. And then to thallium sulfate. The tetraphenylborates are insoluble. These experiments point to a resemblance between potassium and thallium.Video 7 A comparison of some reactions of thallium(I) and potassium ions.Interactive feature not available in single page view (see it in standard view).Like potassium, thallium forms, unusually, a soluble, alkaline carbonate and hydroxide, Tl2CO3 and TlOH. Both ions also form a precipitate with the addition of a solution containing the tetraphenylborate anion, BPh4− where Ph = phenyl.
4.1.5 The inert pair effect
Thallium is a Group 13 metal like aluminium, so why in its compounds is the +1 oxidation number prominent, such as in water the Tl+(aq) ion?
Consider first Group 2, magnesium is less readily oxidised than the metals beneath it. One sign of this is the less negative values of 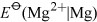 compared with those of the elements below it.
compared with those of the elements below it.
Table 5 compares values for Group 2 and Group 13 metals. Is the trend mentioned above maintained in Group 13?
No the trends are in opposite directions: thermodynamically speaking, aluminium is more readily oxidised to oxidation number +3 than gallium, indium or thallium.
|
| ||
|---|---|---|---|
| Mg | −2.36 | Al | −1.68 |
| Ca | −2.87 | Ga | −0.53 |
| Sr | −2.90 | In | −0.34 |
| Ba | −2.91 | Tl | +0.72 |
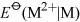
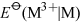
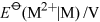
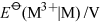
A possible reason for the reversal emerges from a comparison of the first ionisation energies of the Group 2 and Group 13 elements (Figure 20). Generally the ionisation energy:
- drops steeply when a new Period begins and
- increases overall across a Period as the nuclear charge builds up, as seen in Group 2.
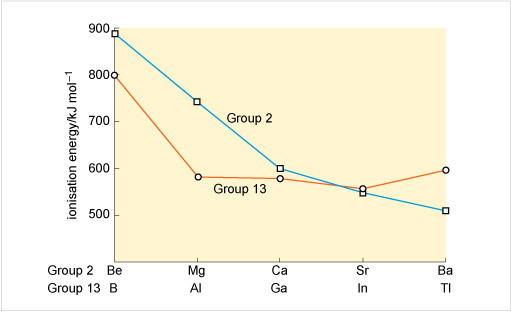
Does Group 13 follow this general trend?
From boron to aluminium, there is the usual drop from the second row to the third row of the Group. Thereafter, the values remain unexpectedly high, most notably at thallium whose first ionisation energy exceeds that of aluminium.
Consider Figure 21. Why might the ionisation energies of gallium, indium and especially thallium be unexpectedly raised relative to those of aluminium?
Aluminium follows immediately after a Group 2 element, but before gallium and indium, d shells must be filled. The ionisation energies are higher for gallium and indium because it takes longer to build up the nuclear charge. The effect is magnified at thallium, where prior filling of 5d and 4f subshells occurs.
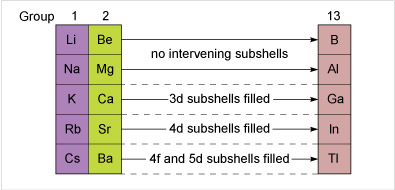
Unexpectedly high ionisation energies for gallium, indium and thallium make conversion of the metals into ions more difficult. They are a major contribution to the greater resistance to oxidation revealed in Table 5. Such effects, however, do not obliterate the strong resemblance of gallium, indium and thallium to aluminium that their presence in the same Group implies. All three elements are metals, which react with fluorine or chlorine to form trihalides, all of which are solids at room temperature.
However the build-up of nuclear charge in the preceding 4f and 5d block elements leaves thallium's ionisation energies higher than expected. The higher oxidation number is therefore harder to attain, and the state most stable to oxidation or reduction is +1.
Determine the electronic configuration of Tl+.
[Xe] 4f14 5d10 6s2; the outer 6p electron has been lost, leaving two outer electrons in a full 6s shell.
The inert pair effect refers to the emergence at the bottom of Groups 13-15 of a stable lower oxidation number two fewer than the Group number. This is so called because the outer electronic configuration of the ion is a filled s2 subshell, which is presumed to be hard to remove during oxidation.
Watch Video 8 and determine which compounds aluminium commonly forms upon reaction with the halogens.
Download this video clip.Video player: Video 8Transcript: Video 8 The reaction of aluminium with some halogens.
End transcript: Video 8 The reaction of aluminium with some halogens.NARRATORAluminium forms solid trihalides when heated with the halogens. Here, we're heating aluminium in a stream of chlorine gas. An exothermic reaction produces white aluminium chloride. Adding aluminium foil to bromine produces aluminium tribromide in a spectacular reaction.The iodide has the same structure. Here, we mix aluminium with iodine. A drop of water dissolves a little of the iodine and allows the reactants to mix. The heat of the reaction is enough to sublime iodine, hence the purple vapour.Video 8 The reaction of aluminium with some halogens.Interactive feature not available in single page view (see it in standard view).Aluminium forms trihalides such as AlCl3.
The inert pair effect increases down the Group: AlCl, AlBr and AlI do not exist at room temperature, but the corresponding compounds of gallium and indium can be made by cooling a heated mixture of the metals and their trihalides:
They all, however, decompose in water, either evolving hydrogen or disproportionating to the metal and M3+(aq). Only in the case of thallium does a long-lived M+(aq) ion exist.
The inert pair effect is a relative phenomenon; consider whether it is also apparent if you study the two ionisation energies of the valence shell s electron pair.
For example, the atoms of the Group 2 elements have configurations of the type [FIS] ns2, (FIS refers to a filled inner shell) and in the +2 oxidation state, their ionic configuration is [FIS]. For a Group 2 element, the process
involves the loss of the outer s pair, and its enthalpy change is (I1 + I2), i.e. the sum of the first and second ionisation energies.
Rewrite this last sentence so that it applies to the loss of the outer s pair in Group 13.
For a Group 13 element, the process (Equation 51) involves the loss of the outer s pair, and its enthalpy change is (I2 + I3), the sum of the second and third ionisation energies.
M+(g) = M3+(g) + 2e−(g)(Equation 51)
Figure 22 shows the change in these ionisation energy sums as one descends Groups 2 and 13. To make the comparison easier, the scales for each plot have been adjusted so that the slopes of the lines between Be and Mg, and between B and Al, are the same.
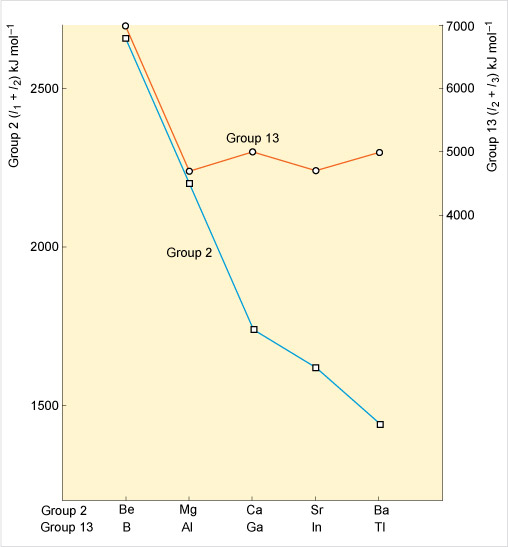
In Figure 22 do Groups 2 and 13 elements follow the trend you would expect?
Descending Group 2, the ionisation energy falls as expected because the outer shell becomes more remote from the nucleus: it gets progressively easier to remove the s pair.
Whilst in Group 13, the slope of the decrease changes abruptly at aluminium. Indeed, between Al and Ga, and In and Tl, there are increases. Unlike Group 2, it gets progressively harder to remove the s pair upon descending Group 13 due to an increasing nuclear charge from filling first the d subshell before Ga, In and Tl and also the f subshell before Tl. Consequently in Group 13, the +1 oxidation state becomes much more stable moving from Al to Tl.
4.2 Home water filters
Many people use home water filters. These effectively remove both hardness and impurities that may alter the taste of tap water, such as residual chlorine. Have you ever wondered what is inside a refill cartridge (Figure 23)?

- The filter contains an adsorbent material that adsorbs a wide range of compounds, including organic molecules such as pesticides.
- Activated carbon is commonly used as it has a high surface area.
- The structure of activated carbon is similar to graphite. It differs in having a smaller particle size and lower crystallinity.
- Also the edge of the graphite-like sheets (Figure 24) have been oxidised which helps improve its absorbent properties.
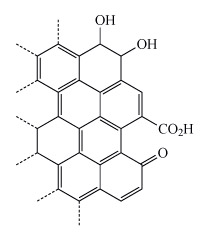
Why might the additional functional groups from oxidation make activated carbon a better adsorbent than graphite with a similar surface area?
The additional functional groups enable additional intermolecular interactions between the adsorbent and the pollutant. For example, the hydroxyl groups can hydrogen bond with anions such as chloride or nitrate. Similarly the carboxylic acid and carbonyl groups can coordinate cations such as calcium or magnesium.
Activated carbon is also widely used as an adsorbent to treat industrial wastewater.
Summary of Section 4
- Water hardness is often due to the presence of dissolved calcium and magnesium salts in the water, such as Ca(HCO3)2.
- Aluminium oxide and hydroxide are unusual in being amphoteric. This means that they dissolve in and neutralise both alkali and acidic solutions. For example: Al(OH)3(s) + 3H+(aq) = Al3+(aq) + 3H2O(l)(Equation 39)Al(OH)3(s) + OH−(aq) = [Al(OH)4]−(aq)(Equation 40)
Aluminium sulfate is used to remove fine particles from water, wherein the following reaction occurs:
Al3+(aq) + 3HCO3−(aq) = Al(OH)3(s) + 3CO2(g)(Equation 42)as the aluminium hydroxide precipitates it encourages the fine particles to settle out.
- In the body, aluminium(III) ions are toxic by interfering with the binding of magnesium(II) to ATP during its hydrolysis.
- Thallium(I) is poisonous by replacing potassium ions, which have a similar ionic radius, in the body and disrupting normal function.
- Although thallium is in Group 13 its common oxidation state is +1 due to the inert pair effect. The inert pair effect is also observed in Groups 14, 15 and 16.
- Activated carbon is a common adsorbent material in water filters that removes anionic, cationic and organic pollutants.
Conclusion
This free course provided an introduction to studying Science. It took you through a series of exercises designed to develop your approach to study and learning at a distance, and helped to improve your confidence as an independent learner.
Glossary
- Amphoteric
- An amphoteric compound dissolves in and reacts with both acidic and alkaline solutions.
- Bronsted-Løwry
- The Bronsted-Løwry theory of acids and bases states that an acid is a substance from which a proton can be removed and a base is a substance that can accept a proton from an acid.
- Anions
- A negatively charged ionic species.
- Catena-
- A prefix distinguishing a linear condensed anion.
- Cations
- A positively charged ionic species.
- Chelating
- A chemical species (a ligand) that contains two or more atoms with lone pairs of electrons that may bind to a metal ion. Their complexes show enhanced stability compared to ones where each ligand only binds at one position to the metal ion.
- Coordination number
- The number of roughly equidistant nearest-neighbour atoms that surround another atom.
- Cyclo-
- A prefix to denote a cyclic condensed anion.
- Eutrophication
- The term given to the death of life forms in bodies of water, such as lakes due to excess phosphate and nitrate fertilisers in the water.
- General formula of oxoacids
- In an oxoacid there may be one or more terminal oxygen atoms, so the general formula of oxoacids can be applied, A(O)t(OH)n, where t can equal 0. For example, sulfuric acid, H2SO4, is written as S(O)2(OH)2 (where t = 2, n = 2).
- Hard and soft acids and bases
- Hard acids and hard bases have low polarisability (they are small compact atoms or ions), whereas soft acids and soft bases are more polarisable (they are larger atoms and ions). Hard acids include the cations of the alkali metals, the alkaline earths and the first-row transition metals in high oxidisation states, and hard bases include ammonia and the fluoride ion. Soft acids include the copper(I), silver(I) and mercury(II) ions, and the larger halogens. Soft bases include the cyanide ion, phosphines and the iodide ion. Hard acids tend to bond most effectively to hard bases, and soft acids to soft bases.
- Hydrophilic
- Literally, 'water-loving'. A hydrophilic compound, such as an ionic species, will dissolve in water.
- Hydrophobic
- Literally, 'water-hating'. A hydrophobic compound, such as a non-polar organic compound, will be totally immiscible with water.
- Inert pair effect
- The tendency of the typical elements at the bottom of Groups 13-16 to adopt an oxidation number two less than the Group number.
- Negatively charged ions
- A negatively charged ionic species, also referred to as anions.
- Nitrogen fixation
- A collective term for all the natural processes that convert atmospheric N2 into useful compounds such as ammonia and nitrates.
- Oxidisation number
- To assign the oxidisation numbers, or oxidisation states, within a compound we imagine that the shared electron pairs in covalent bonds between different elements are completely transferred to the more electronegative of the two elements. For example, in sodium chloride, sodium has an oxidisation state of +1 and chlorine has has an oxidisation state of -1. In covalent compounds, the sign reflects the relative electronegativity of the different elements, for example in water, H2O, the H is +1 and the O is -2.
- Polybasic
- A polybasic acid contains more than one ionisable proton, H+ ion.
- Polyprotic
- A compound that contains several ionisable protons, H+. For example sulphuric acid is a polyprotic acid.
- Positively charged ions
- A positively charged ionic species, often referred to as a cation.
- Redox
- A reaction in which oxidation and reduction occur.
- Stoichiometry
- This can have either of two meanings, depending on the context in which the term is used. In inorganic chemistry, with NaCl, for instance, if there are n atoms in the crystal lattice, then there are n octahedral holes, and so the Na : Cl ratio will be 1 : 1. The stoichiometry in this case is 1 : 1. In the context of chemical reactions, the stoichiometry represents the relative amounts of reactants and products taking part, as expressed by the balanced chemical equation.
- VSEPR theory (valence-shell electron-pair repulsion theory)
- A theory for predicting molecular shapes by postulating that electron pairs in molecules will be kept as far away from each other as possible.
References
Further reading
Acknowledgements
This course was written by Simon Collinson with contributions from Rob Janes, Michael Gagan, Craig Williams, Joan Mason, Charles Harding and John Baxter.
Course image: Chafer Machinery in Flickr made available under Creative Commons Attribution-NonCommercial-ShareAlike 2.0 Licence.
The material acknowledged below is Proprietary and used under licence (not subject to Creative Commons Licence). Grateful acknowledgement is made to the following sources for permission to reproduce material in this course:
Figures
Figure 10 Dr Gene Feldman, NASA GSFC/Science Photo Library;
Figure 15 Building Research Establishment;
Every effort has been made to contact copyright owners. If any have been inadvertently overlooked, the publishers will be pleased to make the necessary arrangements at the first opportunity.
Except for third party materials and otherwise stated in the acknowledgements section, this content is made available under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 Licence.
Don't miss out:
If reading this text has inspired you to learn more, you may be interested in joining the millions of people who discover our free learning resources and qualifications by visiting The Open University - www.open.edu/ openlearn/ free-courses