

4 Physiological adaptations – molecules and cells
4.1 Scientific approaches
Even after many years of research, the phenomenon of hibernation continues to be a mystery to scientists. Despite coming nearer to an understanding of how and why it happens, some fundamental questions remain unanswered. Is there a genetic basis underlying the evolutionary predisposition of animals to hibernate, given its occurrence in many groups of vertebrates and invertebrates? Is the problem of metabolic adaptation in cells separate from thermal regulation which occurs throughout the organism? In Section 4 we will attempt to answer these questions, starting here by looking at scientific approaches.
Faced with an exploration of the unknown, rather than simply testing an existing hypothesis, research can adopt two different approaches in attempting to associate molecular events with physiological functions.
The analytical approach seeks to identify all the significant changes that accompany a specific physiological adaptation and then seeks to explain these changes. For example, differences in the patterns of expression of a number of genes that manifest themselves during hibernation can be identified against a background of no change. The processing of very large numbers of genes in this way became possible in the late 1990s with the advent of DNA chip technology. This powerful approach can provide information about the state of expression of thousands of genes in each tissue (Box 2).
Such an analytical approach has led to the identification of a few genes in a range of species whose expression is either increased or decreased during hibernation (Table 4).The table shows that some of the genes are associated with the metabolic, respiratory or control functions that you might predict would be linked to the maintenance of, or recovery from, hibernation. The links revealed in this kind of analysis sometimes appear indirect or tenuous. The benefit of this ‘needle-in-the-haystack’ approach, however, is that it reveals changes in expression patterns in a handful of proteins amongst a very large number which do not undergo such changes – even those that might be predicted to do so. Genetic analysis has revealed significant changes in the expression of genes whose function is not known or not directly related to hibernation (for example, the HT20 gene family in Table 4).
| Gene | Tissue | Change in expression | Possible function |
|---|---|---|---|
| apoferritin | liver | increase | iron storage protein – increases availability of iron for cytochromes and haemoglobin |
| c-fos | brain | redistribution+ | rapid-response gene – coordinates reaction to physiological change |
| genes for fatty acid binding proteins | BAT | increase | preparation for rapid thermogenesis during arousal |
| genes for glyceraldehyde phosphate dehydrogenase | liver, muscle | decrease | reduces glycolysis |
| HT20 family | muscle, WAT | decrease | function unknown – all related structurally to protease inhibitor α1 anti-trypsin |
| genes for pyruvate dehydrogenase | heart, skeletal muscle | increase | depresses metabolism by preventing pyruvate kinase conversion into acetyl CoA |
Footnotes
+ c-fos expression undergoes a decrease in the hypothalamus during hibernation and a redistribution within specific neuronal nuclei on arousal.
Box 2: DNA ‘microarrays’ for the study of gene expression
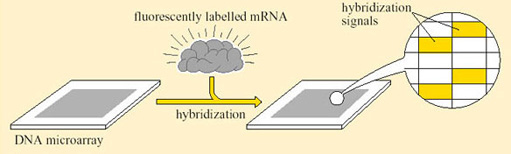
To elucidate the entire mRNA content of a cell, or to study all the genes that are being actively transcribed at any one time, every protein-coding gene in the genome would have to be analysed. The enormity of this task can be appreciated only by considering the number of genes that are activated in a single biological process. For example, during the transition from aerobic to anaerobic respiration in cells of the yeast S. cerevisiae, changes in the expression of 1740 genes have been recorded. This large change in gene expression in S. cerevisiae has been analysed using the technique known as DNA microarray (Figure 22).
The design of DNA ‘chips’ allows many hybridization experiments to be performed in parallel. A DNA chip is a very thin layer of silicon, 2 cm2 or less in area, carrying a large number of DNA probes, a microarray, each with a different sequence and each at a defined position on the chip. The probes can be short oligonucleotide sequences, and can be spotted onto the silicon using high-speed robotics to form a microarray, which is then incubated with the labelled target to allow hybridization to take place. To determine which oligonucleotides have hybridized to the target, the surface of the chip is scanned and the positions at which the signal emitted by the label is detectable are recorded.
Studies on cancerous tissue using this technique discovered genes whose expression patterns differed significantly when normal colon epithelial cells were compared with colon cancer cells. In brief the method was as follows (Figure 23). Messenger RNA preparations were made from cancerous and normal cells. Each preparation was then labelled with a radioisotope attached to a fluorescent marker and allowed to hybridize to a microarray containing probes for several thousand human genes. The hybridization signal associated with each gene was then used to assess the particular mRNA in each preparation. In pancreatic cancer cells, about half of these genes also showed abnormal expression levels. The implication of this study is that some genes are abnormally expressed in more than one type of cancer while others are expressed more specifically. DNA microchip technology has been one way to enable such studies of gene expression.
Question 10
How can we attach relevance to an observed change in gene expression, and hence predicted changes in the biosynthesis of the protein which the gene encodes?
Answer
First, the nucleotide sequence of each gene is compared with those in the genomic database for the species. High levels of sequence homology (similarity) enable us to predict functional analogies with these genes on the basis of predicted protein structure, spatial (tissue location) and temporal (daily or seasonal) expression patterns. If such a search fails to establish any homology with any known gene, it is necessary to establish a function without precedent for the new protein. The most valuable evidence frequently comes from observations on loss of physiological function when one or both copies of the gene are deleted from the genome. Hypotheses on the function of the protein may then be tested by determining the proteins and subcellular structures with which it interacts in order to fulfil its function.
The analytical approach has indicated that different tissue-specific genes are activated or repressed in the brain and peripheral organs of hibernating animals. The genes encode pre-identified proteins which are linked to the role of each tissue in hibernation (i.e. reactive control in the brain, metabolic adaptation in liver muscle and adipose tissue) plus some proteins whose function is unclear.
The targeted approach seeks to investigate a possible role for molecules already suspected of participating in physiological regulation. An example is the enzyme arylalkylamine-N-acetyltransferase (AA-NAT). This enzyme catalyses the rate-limiting step in the production of a hormone, melatonin, from the pineal gland. Melatonin has the capacity to re-set circadian rhythms in a variety of physiological processes. Entry into hibernation requires neural and endocrine control systems that govern the normal day/night (circadian) cycles of metabolism and T b, to be overridden.
Question 11
What would you expect analysis of gene expression to show?
Answer
Expression of AA-NAT, leading to elevated biosynthesis of melatonin, should increase just prior to onset and during hibernation. Circadian rhythms would then be interrupted whilst the animal is torpid.
This prediction was supported by studies using 13-lined ground squirrels. Messenger RNA for AA-NAT protein biosynthesis increased in the brains of hibernating animals as expected – an accepted indication that the enzyme's activity had increased (Figure 24) (Yu et al., 2002).

In a second example, researchers proposed a working hypothesis that the uptake of fatty acids by hibernating tissues, which is normally under tight control, should be deregulated in preparation for the task of storing large reserves of lipid. They expected to see changes in the activity of a key enzyme, acetyl CoA carboxylase. This enzyme catalyses the formation of malonyl CoA, a potent and major inhibitor of mitochondrial fatty acid uptake. In studies on Richardson's ground squirrel, the hypothesis was proved right. Acetyl CoA carboxylase in heart muscle was much reduced prior to and during hibernation, so allowing more fatty acid uptake by mitochondria in cardiac muscle.
Box 3: Regulated/regulating enzymes that influence RQ value and energy fuel selection in adipose tissue and muscle
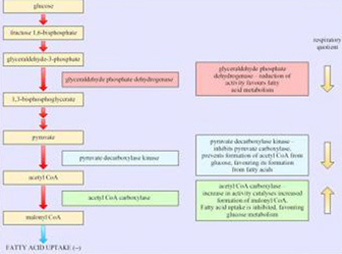
Gene expression research has provided evidence for the enzymes that play an important role in determining the choice of respiratory fuel in adipose tissue and hence the respiratory quotient (RQ; see Box 3). As we will see in the next section, changes in energy sources are a characteristic of hibernators.