

2.6 A gene ‘for’ obesity?
So far we have mostly emphasized the way in which different environmental factors may affect body weight and provide a partial explanation of both individual cases of obesity and the increase in average body weight that has been so clearly documented in both North America and Western Europe during the last two decades. There is also marked individual variability in body weight. For example, any weight between about 58 and 78 kg would be regarded as ‘desirable’ for a person of height 1.77 m.
Activity 28
Why might this range of body weight be regarded as desirable for someone with a height of 1.77 m?
Answer
In Table 4 you will find the range of BMIs regarded as desirable. The lower end of the desirable range is shown as 18.5, which, when substituted into the formula for the BMI of a person of 1.77 m in height, gives a body weight of 57.96 kg. The upper end of the desirable range is 25, giving a weight of 78.32 kg for persons of this height.
Clearly the actual range of body weights found in human populations is very much greater than the desirable range, and the environmental factors discussed earlier may explain a substantial part of this variation between individuals. But it is also possible that some of this variation depends on specific genetic differences between individuals.
There is considerable evidence to suggest that body weight is heritable to some extent. In a classic study, Albert Stunkard and his colleagues (Stunkard et al., 1986) investigated the relationship between adult body weight of 540 adopted Danish adults and the weights of their biological and adoptive parents. The results were clear cut. There was a strong relationship, which extended from individuals who were very thin through to those who were obese, between the weight of adoptees and the weight of both their biological father and mother. There was no relationship between an adoptee's weight and the weight of either adoptive parent. The idea that variability in body weight may be partly under genetic influence also fits with older studies suggesting that different racial groups may have differing propensities to obesity and type 2 diabetes. In 1962, Neel reviewed data suggesting that certain human populations, including some South Pacific Islanders and North American Indians, may have a genotype that promotes increased fat storage. This genotype represents a survival advantage in times of famine, but can predispose these populations to obesity and type 2 diabetes in typical Westernized societies (see Bindon and Baker, 1997). This is usually known as the 'thrifty genotype’ hypothesis. It is important to note that this hypothesis is not suggested as a universal, or even a common, reason for obesity, but one that applies just to these specific groups.
Activity 29
How would you characterise the major difference between the thrifty genotype and the thrifty phenotype hypotheses of obesity?
Answer
The thrifty phenotype hypothesis emphasizes the way in which an environmental trigger (poor fetal nutrition) can alter the course of development so as to make obesity more likely when the environment is appropriate. By contrast, the thrifty genotype hypothesis suggests that genetic differences between (racial) groups of individuals makes obesity a more likely consequence in some of those groups when they are exposed to an energy-dense modern diet.
Recent advances in our understanding of both molecular genetics and the hormonal and neurotransmitter controls of feeding have led to specific explanations of some causes of obesity.
Leptin is a hormone produced by adipose tissue that acts on specific nerve cells in the brain stem and hypothalamus to inhibit feeding behaviour. It was first identified because a very obese strain of mice was discovered to have a mutation in the gene that codes for leptin. Subsequently, a very small number of human families have been identified in which some individuals also have a mutation in the gene that codes for leptin. These mutations are often of the ‘frame-shift’ type and lead to a complete loss of function in the resulting protein. Humans with such mutations overeat and become very obese. If, as children, attempts are made to restrict their food intake, they may become aggressive and difficult to manage. Treatment with leptin leads to normalization of both appetite and body weight. However, the administration of leptin turns out to be very much less effective in the great majority of morbidly obese individuals. They typically already have very high leptin levels but have lost the normal physiological and behavioural responses to the hormone. Thus leptin has not, so far, proved to be the ‘magic bullet’ that will treat the human obesity epidemic.
In the hypothalamus, leptin acts on two groups of nerve cells, both of which influence feeding behaviour but with opposite actions. One set of cells uses neuropeptide Y (NPY) as their neurotransmitter and activation of these cells tends to increase appetite (Figure 11). The second set of cells uses ![]() -melanocyte-stimulating hormone (
-melanocyte-stimulating hormone (![]() -MSH) as a neurotransmitter. The
-MSH) as a neurotransmitter. The ![]() -MSH acts on a subtype of melanocortin receptor known as the MC4 receptor located on other hypothalamic neurons. Activation of the
-MSH acts on a subtype of melanocortin receptor known as the MC4 receptor located on other hypothalamic neurons. Activation of the ![]() -MSH-containing cells is associated with a reduction in feeding behaviour. As you would predict, mice which have been genetically manipulated so that the MC4 receptor is non-functional overeat and are obese. The functional receptor works by producing a second messenger in the postsynaptic cell when
-MSH-containing cells is associated with a reduction in feeding behaviour. As you would predict, mice which have been genetically manipulated so that the MC4 receptor is non-functional overeat and are obese. The functional receptor works by producing a second messenger in the postsynaptic cell when ![]() -MSH is released presynaptically. The production of second messenger is said to be coupled to
-MSH is released presynaptically. The production of second messenger is said to be coupled to ![]() -MSH binding.
-MSH binding.
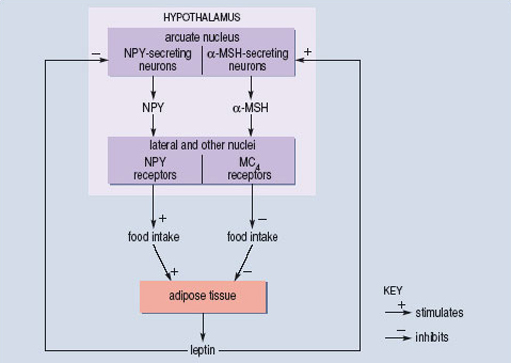
O'Rahilly and colleagues, working at Cambridge University (Farooqi et al., 2003), have shown that mutations in the human MC4 receptor may account for about 5% of cases of morbid obesity. A number of different point mutations have been isolated, and are associated with different degrees of obesity. The extent to which each mutation interferes with the coupling between ![]() -MSH binding to the receptor and the production of second messenger has also been determined. There is a good correlation between the two measures. Mutations that are associated with the greatest degree of obesity are those in which coupling is least efficient. So, for these cases, we have a reasonably complete understanding of the way in which single mutations in a gene may lead to changes in a complex behavioural trait such as feeding and then go on to influence the long-term regulation of body weight. But even here the statistical correlation is far from perfect, which means that the genetic variation explains only a part of the individual variation in body weight.
-MSH binding to the receptor and the production of second messenger has also been determined. There is a good correlation between the two measures. Mutations that are associated with the greatest degree of obesity are those in which coupling is least efficient. So, for these cases, we have a reasonably complete understanding of the way in which single mutations in a gene may lead to changes in a complex behavioural trait such as feeding and then go on to influence the long-term regulation of body weight. But even here the statistical correlation is far from perfect, which means that the genetic variation explains only a part of the individual variation in body weight.
However, in most cases inheritance of obesity is not consistent with a single gene change. If you look back over the different examples that we have discussed, you can see that this is exactly what would be expected. Even though obesity can only reflect greater energy input than output, the imbalance may arise in many different ways. Environmental factors may play an important role, as may early programming of physiology following fetal undernutrition. Genetic factors may also play a critical role in some cases. But to specify the relative importance of these different factors for any particular individual is always likely to be very difficult. However, our increasing understanding of the molecular genetics of obesity can be very positive in specific cases. For example, for those few individuals who are leptin deficient, administration of synthetic leptin provides good treatment. Our increased understanding also makes it clear that to label young individuals with this type of deficiency as gluttons who lack self control, and to characterize their parents as inadequate for failing to control their child's diet and weight is completely inappropriate.