
5.2 Thalassaemia
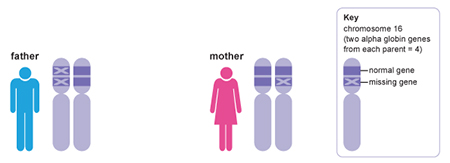
Thalassaemias are a group of inherited autosomal recessive disorders that cause anaemia because of the decreased or absent synthesis of a globin chain (Muncie and Campbell, 2009). Alpha thalassaemia is the result of either deficient or absent production of the alpha globin chain, which is then replaced by extra beta globin chains. Production of the alpha globin protein is slightly more complicated because it is controlled by two genes, both located on chromosome 16. This means that disease susceptibility is dependent on the inheritance pattern of four alleles – two inherited from the mother and two from the father. Alpha thalassaemia is usually due to the deletion of one of these alleles and the severity of the disease corresponds to the number of deletions:
- one deletion is silent and asymptomatic
- two deletions result in mild anaemia
- three deletions cause haemoglobin H disease and moderate to severe anaemia
- four deletions cause alpha thalassaemia major, a fatal condition.
Activity 10 Inheritance pattern of alpha thalassaemia
In this activity, you will predict the phenotype and pattern of inheritance of alpha globin genes in a family affected by alpha thalassaemia. Click below to reach the full activity.
Beta thalassaemia results from deficient or absent production of the beta globin chains, leading to excess alpha chains in the Hb molecules (Figure 15).
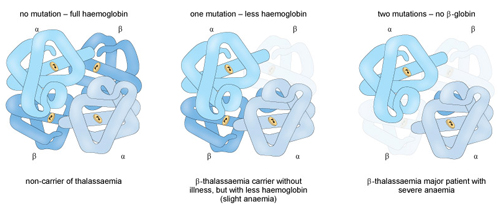
Unlike alpha thalassaemia, beta thalassaemia is usually due to a point mutation (more than 200 of which have been identified to date) in the gene that codes for beta globin. Again, the degree of disease symptomology is dependent on how many beta globin chains are functional. Beta thalassaemia minor is asymptomatic whereas beta thalassaemia major causes growth retardation, skeletal abnormalities and jaundice, and requires lifelong blood transfusions to treat.
The overall effect of either alpha or beta thalassaemia is haemolysis, the rupture and destruction of the erythrocytes. Because of this, people with thalassaemia are at risk of developing pulmonary hypertension, a higher than normal pressure in the arteries that carry blood to and from the lungs. This can cause dizziness, shortness of breath and damage to the heart.
Question 11 Hb mutations
Why do you think Hb mutations that cause potentially fatal anaemias continue to exist in the human genome?
Answer
The rates of both sickle cell anaemia and thalassaemias are higher in people of African, Southeast Asian and Mediterranean descent. It is not a coincidence that these are also regions where the malaria parasite is highly prevalent. Heterozygous carriers of the HbS gene or thalassaemia mutations are less likely to be infected with the Falciparum malaria parasite than people with the normal copies of those genes. Malaria can cause serious illness and over one million people die from the infection every year. Therefore, mutations in Hb lead to a trade-off between increased risk of anaemia and decreased risk of death from malaria. Malaria is a good example of how parasites (and other infectious organisms) exert evolutionary pressure on the human genome to adopt multiple polymorphisms that protect against severe disease.