Cell signalling
Use 'Print preview' to check the number of pages and printer settings.
Print functionality varies between browsers.
Printable page generated Friday, 26 April 2024, 2:28 AM
Cell signalling
Introduction
Even the simplest organisms can detect and respond to events in their ever-changing environment. Similarly, within a multicellular organism, cells are surrounded by an extracellular environment from which signals are received and responded to. Extracellular events are decoded and transmitted to relevant parts of individual cells by way of a series of activation/deactivation steps involving many intracellular molecules. This relay of information along molecular pathways is called signal transduction; it is sometimes also simply referred to as ‘signalling’.
The molecular models shown in this chapter were produced using the Brookhaven protein data base (pdb) files indicated in the figure legends. These files can be downloaded, viewed and manipulated using a suitable molecular viewing programme, such as Viewerlite tm.
This OpenLearn course provides a sample of Level 3 study in Science.
Learning outcomes
After studying this course, you should be able to:
define and use each of the terms printed in bold in the text
understand the basic principles of signal transduction mechanisms, in particular the concepts of response specificity, signal amplitude and duration, signal integration and intracellular location
give examples of different types of extracellular signals and receptors, and explain their functional significance
describe the mechanisms by which different receptors may be activated by their respective ligands
describe and give examples of the structure and properties of the major components of signal transduction pathways.
1 General principles of signal transduction
1.1 Introduction
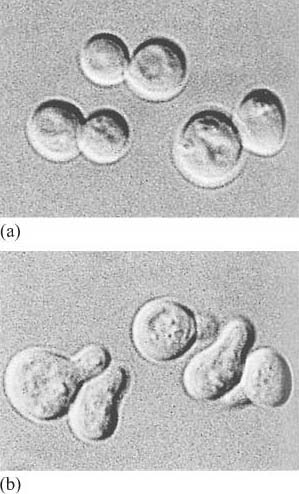
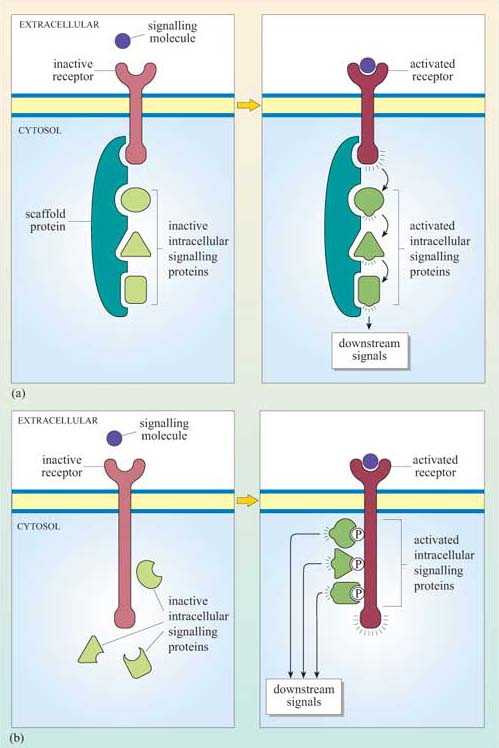
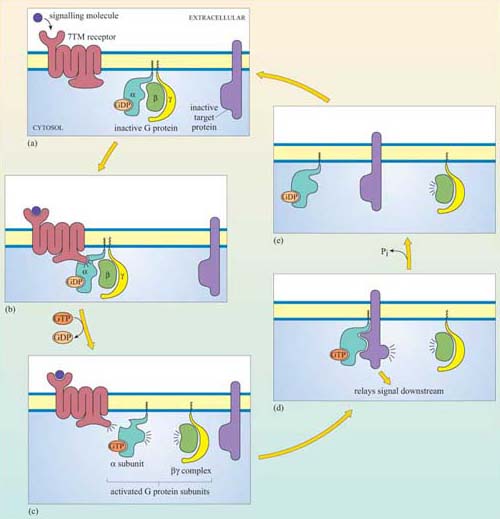
The fundamental principles of signalling can be illustrated by a simple example in the yeast S. cerevisiae (Figure 1). In order to sexually reproduce, a yeast cell needs to be able to make physical contact with another yeast cell. First, it has to ‘call’ to yeast cells of the opposite mating type. It does this by secreting a ‘mating factor’ peptide, an extracellular signal, which can also be called an ‘intercellular signal’. Yeast mating factor binds to specific cell surface receptors on cells of the opposite mating type, and the signal is relayed into the target cell via a chain of interacting intracellular signalling molecules, which switch from an inactive (Figure 2a) to an active state (Figure 2b). Signalling molecules are said to be upstream or downstream of other components of the pathway (this terminology should not be confused with that used to describe the structure of genes in relation to transcription). Ultimately, signalling molecules activate target effector proteins (an effector in this context is a molecule that carries out the cellular response(s) of the signalling pathway). In the yeast, signal transduction to mating factor ultimately stops the target cell proliferating, and induces morphological changes which result in the formation of protrusions towards the cell that releases the mating factor (Figure 1b). The morphological changes are a response to the signal. The two cells can then make physical contact with each other, and mating can ensue.
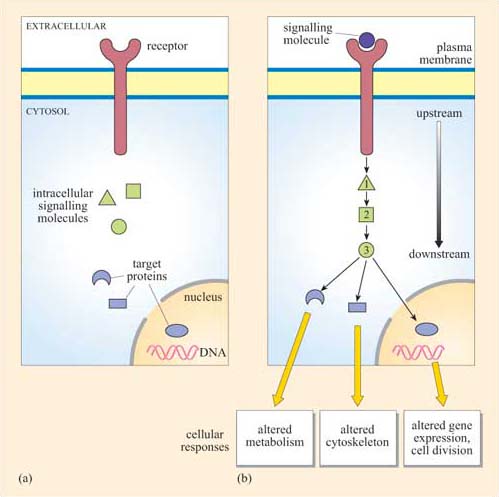
Which signal pathway molecule(s) can be said to be upstream and downstream of molecule 2 in Figure 2b?
The receptor and intracellular signalling molecule 1 are upstream; both intracellular signalling molecule 3 and the target proteins are downstream.
Signalling in multicellular organisms is a complex process, in which many millions of highly specialized cells may need to act in a coordinated fashion. Cells may need to respond to several signals at once, and different cells may need to respond to the same signal in different ways. All this is made possible because the mechanism for detection of a signal is not directly coupled to the response, but is separated by a chain of signalling events, such as that shown in outline in Figure 2b. This principle allows signalling systems to be highly flexible. Examples of this flexibility are:
the same type of receptor can be coupled to different signalling pathways in different cell types;
the signal can be amplified (or damped down) as it travels along the signalling pathway;
it can switch on multiple pathways, leading to several cellular responses in diverse regions of the cell;
information can be processed from several different receptors at once to produce an integrated response.
Most of this is made possible by protein–protein interactions and protein regulatory mechanisms.
Despite this complexity, the basic model of signal transduction set out in Figure 2 holds true for most intracellular signalling pathways across species, and often the signalling molecules themselves are highly conserved. For example, there is a high degree of homology between the major proteins in the yeast mating factor signalling pathway and the human mitogen-activated protein (MAP) kinase growth signalling pathway.
A mitogen is an extracellular molecule that induces mitosis in cells.
In this course, we shall guide you through the signalling network, firstly by introducing you to the kinds of molecules involved in signal transduction and the general principles employed by cells. Then we shall go into greater detail to show you exactly how the key molecular players operate including receptors and intracellular signalling molecules. In the final section, we shall consider specific examples of signal transduction pathways regulating cellular responses involved in glucose metabolism in different cell types.
1.2 Extracellular signals can act locally or at a distance
First we shall consider the general types of intercellular signalling mechanism within multicellular organisms (Figure 3). Broadly speaking, cells may interact with each other directly, requiring cell–cell contact, or indirectly, via molecules secreted by one cell, which are then carried away to target cells.
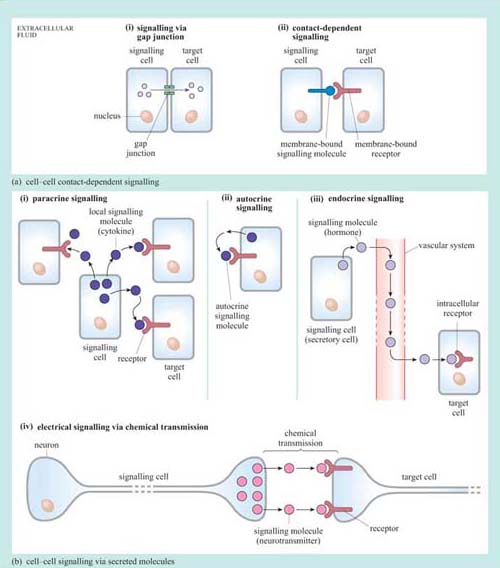
1.2.1 Cell–cell contact-dependent signalling
In some instances, cells may communicate directly with their immediate neighbour through gap junctions (Figure 3a). Communication via gap junctions partially bypasses the signalling model we have outlined above in Figure 2. Gap junctions connect the cytoplasm of neighbouring cells via protein channels, which allow the passage of ions and small molecules (such as amino acids) between them (as an example, gap junctions allow the coordinated contraction of cardiac muscle cells).
Alternatively, cells can interact in a ‘classic’ signalling manner, through cell surface molecules, in a so-called contact-dependent way (Figure 3a (ii)). Here the signalling molecule is not secreted, but is bound to the plasma membrane of the signalling cell (or may even form part of the extracellular matrix), and interacts directly with the receptor exposed on the surface of the target cell. This type of signalling is particularly important between immune cells, where it forms the basis of antigen presentation and the initiation of the immune response, and also during development, when tissues are forming and communication between cells and their neighbours is paramount in deciding between cell fates such as proliferation, migration, death or differentiation.
1.2.2 Cell–cell signalling via secreted molecules
Extracellular signalling molecules are all fairly small, and are easily conveyed to the site of action; they are structurally very diverse. The classification and individual names of these mainly water-soluble mediators often reflect their first discovered action rather than their structure. So, for example, growth factors direct cell survival, growth and proliferation, and interleukins stimulate immune cells (leukocytes). However, to complicate matters further, they often have different effects on different cells, and so sometimes their names can appear confusing. Signalling via secreted signalling molecules can be paracrine (acting on neighbouring cells), autocrine (acting on the cell that secretes the signalling molecule), endocrine (acting on cells that are remote from the secreting cell) or electrical (between two neurons or between a neuron and a target cell).
i.In paracrine signalling (Figure 3b (i)) water-soluble signal molecules called cytokines diffuse through the extracellular fluid and act locally on nearby cells. This will usually result in a signal concentration gradient, with the cells in the local area responding differentially to the extracellular signalling molecule according to the concentration they are exposed to (this is an important strategy in development). In order to keep the effect contained, signalling molecules involved in paracrine signalling are usually rapidly taken up by cells or degraded by extracellular enzymes. An example of paracrine signalling involves the gaseous molecule nitric oxide (NO), which, among other effects, acts by relaxing smooth muscle cells around blood vessels, resulting in increased blood flow. As the NO molecule is small (and diffuses readily) and short-lived (so only having time to produce local effects), it fulfils the requirements for a paracrine signalling molecule perfectly.
ii.Autocrine signalling (Figure 3b (ii)) is an interesting variant of paracrine signalling. In this scenario, the secreted signal acts back on the same cell or group of cells it was secreted from. In development, autocrine signalling reinforces a particular developmental commitment of a cell type. Autocrine signalling can promote inappropriate proliferation, as may be the case in tumour cells.
iii.Endocrine signalling (Figure 3b (iii)) is a kind of signalling in which signals are transmitted over larger distances, for example from one organ, such as the brain, to another, such as the adrenal gland. For long-distance signalling, diffusion through the extracellular fluid is obviously inadequate. In such cases, signalling molecules may be transported in the blood. Secretory cells that produce signalling molecules are called endocrine cells, and are often found in specialized endocrine organs. Blood-borne signalling molecules were the first to be discovered and are collectively known as hormones, though they are chemically very diverse. They include steroid hormones (such as the sex hormones and cortisol), some peptide hormones such as insulin, and modified amines that can also act as neurotransmitters (see below) such as noradrenalin. Steroid hormones are biosynthesized from cholesterol. Because they are water-insoluble, they are transported in the blood by specific carrier proteins and are quite stable (their half-lives can be measured in hours or days). This is in contrast to water-soluble signalling molecules, which are much more prone to degradation by extracellular enzymes. Hence they tend to be short lived and are involved in short-term paracrine signalling.
iv.Electrical signalling (Figure 3b (iv)) via chemical transmission (also called synaptic signalling) is a faster and more specific form of cell–cell signalling. Nerve cells, or neurons, can convey signals across considerable distances to the next neuron in the neuronal network within milliseconds. By contrast, blood-borne messages can only operate as fast as blood circulates, but reach many more cellular targets in different tissues. The transfer of information from one neuron to the next is mediated by complex structures called synapses, which are essentially formed by a presynaptic terminal (neuron 1), a synaptic cleft (the tiny gap between the two neurons) and a postsynaptic membrane (neuron 2). When electrical signals reach the end of a neuronal axon (the thin tube-like part of neurons), molecules released from the axon can cross the physical gap between cells and bind to receptors in the target cell. These signalling molecules are collectively called neurotransmitters. Again, these are a diverse group of compounds, including amino acids such as glutamate, nucleotides such as ATP, and CoA derivatives such as acetylcholine.
In addition to its role as a neurotransmitter, can you recall what other roles ATP may have in cells?
ATP is used in phosphorylation reactions, as an energy currency and as a building block for nucleic acid synthesis. ATP is only one example by which localization and compartmentalization enables the same molecule to be used effectively for diverse purposes. You will encounter other examples later in this chapter.
1.3 Most receptors are on the cell surface
Water-soluble signalling molecules cannot cross the membrane lipid bilayer, but bind to specific receptors embedded in the plasma membrane. The receptors have an extracellular domain that binds the signalling molecule, a hydrophobic transmembrane domain and an intracellular domain.
Binding of a ligand induces a conformational change in the receptor, in particular that of its intracellular region. It is this conformational change that activates a relay of intracellular signalling molecules, ultimately bringing about the appropriate cellular response represented in Figure 2.
Receptors can be classified structurally into single-pass transmembrane receptors (with one extracellular, one transmembrane and one intracellular region) and multipass transmembrane receptors.However, in terms of their signal transduction characteristics, it is easier to distinguish four groups of receptors (Figure 4).
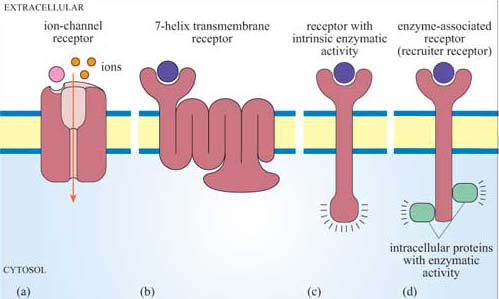
Receptors that also serve as the effector For example, one type of acetylcholine receptor is also an ion channel, and belongs to a family of receptors called ion-channel receptors. In response to acetylcholine, these receptors allow the passage of specific ions, thereby effecting changes in the membrane potential of a cell. Acetylcholine receptors are extremely important in the transmission of electrical signals between excitable cells.
7-helix transmembrane receptors 7TM receptors possess seven membrane-spanning regions, an N-terminal extracellular region and a C-terminal intracellular tail. The mechanism of activation of most 7TM receptors involves coupling to G proteins, and in this case they are also called G protein-coupled receptors (GPCRs). Adrenalin receptors are examples of GPCRs.
Receptors whose intracellular tail contains an enzymatic domain, which are known as receptors with intrinsic enzymatic activity (RIEA) This group includes the receptor tyrosine kinases, involved in the response to many growth factors.
Receptors that require association with cytosolic or membrane-bound proteins with enzymatic activity for signalling These receptors do not have intrinsic enzymatic activity, and have been referred to as enzyme-associated receptors or recruiter receptors (although, strictly speaking, both GPCRs and receptors with intrinsic enzymatic activity also function by recruiting cytosolic signalling molecules, as you will see in Section 3.3).
From an evolutionary perspective (Figure 5), 7TM receptors are of ancient origin and to date have been found in all eukaryotic genomes that have been sequenced, including yeast (a type of 7TM receptor mediates the yeast mating response described in Section 1.2 and Figure 1). Receptors with intrinsic enzymatic activity and many recruiter receptors are found in C. elegans, D. melanogaster and chordates but not yeast, whereas some recruiter receptors, such as T cell receptors that mediate immune responses, are specific to vertebrates (others such as cytokine receptors are specific to chordates, including all vertebrates and some invertebrates such as the sea-squirt).
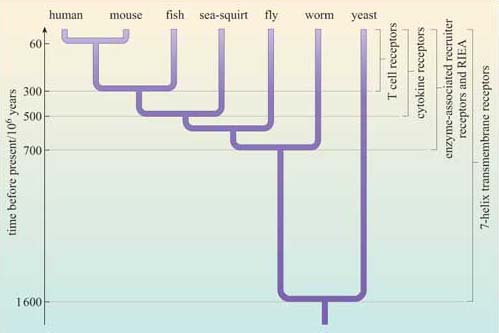
In addition to the four groups of cell-surface receptors shown in Figure 4, another group of receptors function as DNA-binding molecules, and thus regulate gene transcription (these are called receptors with intrinsic transcriptional activity; do not confuse with RIEAs). Some of these receptors are on the cell surface, but most are intracellular (Section 3.5), and require ready access of the ligand to the intracellular compartment.
What sort of ligand might act on an intracellular receptor?
Signalling molecules that can readily diffuse through the cell membrane. These include lipid-soluble compounds such as steroid hormones, and small diffusible molecules such as NO.
1.4 Cellular responses are diverse
Cellular responses can be extremely rapid – for example, the opening of ion channels to effect a change in the membrane potential or the contraction of muscle fibres, which occur within milliseconds of signal reception, or may take minutes, such as whole cell movement, synthesis of new proteins or changes in metabolic activity. There are also longer-term responses, which may be on the scale of hours or even days, such as cell division and programmed cell death. Often several types of response may occur following a single stimulus, in a coordinated manner, and over different timescales.
Within a multicellular organism, a given cell is exposed to many different extracellular signals at any one time. The cell's ultimate response depends on the appropriate integration of these signals and on what cell type it is (for example, only a muscle cell can contract). So, for instance, signal 1 will induce a cell to proliferate but only in the presence of signal 2; in the absence of signal 2, signal 1 will induce the same cell to differentiate. The same signals may produce different responses in different cell types. In the example above, signal 1 might induce cell death in a second cell type. Different cellular responses to an extracellular signal are due at least partly to the specific receptors and intracellular signalling molecules that are active in different cell types. So, not only is the context of the signal vitally important in determining the response but also the type of target cell.
1.5 Signal transduction mechanisms
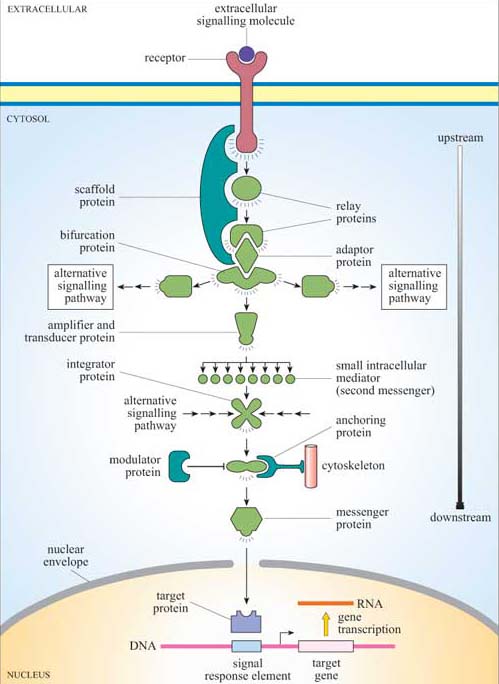
Signalling information has to be transmitted from the receptor in the plasma membrane across the cytoplasm to the nucleus (if gene transcription is the response), the cytoskeleton (if cell movement, or another change to cell morphology, is the response), or various other subcellular compartments. The transmission of a signal must occur in a time-frame appropriate for the cellular response. So, signal transduction needs to take place over both space and time. We have already described a simple signalling model (Figure 2), where a chain of intracellular mediators successively activates the next in the chain until the target is reached. In reality, of course, it is rarely a simple chain, but a branching network, allowing for integration, diversification and modulation of responses (Figure 6). The branched molecular network of activation (and deactivation) of signalling molecules linking receptor activation to the intracellular targets is referred to as a signal transduction pathway (or cascade).
Intracellular signalling molecules have particular properties that allow control of the speed, duration and target of the signal, and may be categorized according to these properties. Broadly speaking, intracellular signalling molecules can be divided into two groups on the basis of molecular characteristics, second messengers and signalling proteins.
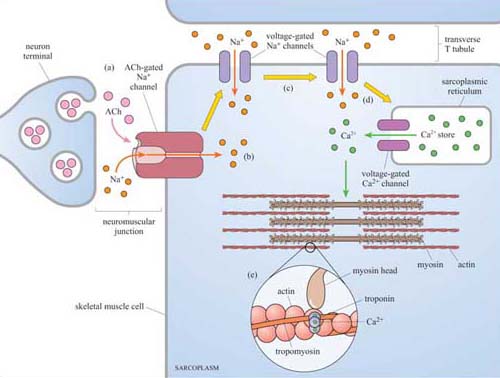
Second messengers are small readily diffusible intracellular mediators, whose concentration inside the cell changes rapidly on receptor activation; in this manner, they regulate the activity of other target signalling molecules. The calcium ion, Ca2+, is a classic example of a second messenger, being released in large quantities in response to a signal (so amplifying the signal) and diffusing rapidly through the cytosol. Ca2+ ions can therefore broadcast the signal quickly to several distant parts of the cell. For example, Ca2+ ions mediate and coordinate contraction of skeletal muscle cells (Figure 7). In general, if a rapid, generalized response is necessary, a second messenger is likely be prominent in the signalling pathway.
Other water-soluble second messengers such as cAMP and cGMP act similarly to Ca2+, by diffusing through the cytosol, whereas second messengers such as diacylglycerol (DAG) are lipid-soluble, and diffuse along the inside of the plasma membrane, in which are anchored various other key signalling proteins.
Second messengers were the first intracellular signalling molecules to be identified; they were so named because hormones or other extracellular signalling molecules were considered the ‘first messengers’. However, the term ‘second messenger’ seems somewhat outdated, since a signalling pathway can easily involve a sequence of eight or more different messengers, and the ‘second messenger’ in question could well actually be acting as, say, the fifth messenger.
Signalling proteins are the large intracellular signalling molecules that generally, but not exclusively, function by activating the next signalling protein in the signal transduction cascade, or by modifying the concentration of second messengers.
Proteins are much larger and generally less mobile than small water-soluble second messengers, so they are not so useful for the rapid dissemination and amplification of a signal. However, proteins are capable of interacting in a highly specific manner with other proteins, they exhibit binding specificity for ligands and for recognition motifs on other molecules, and their activity can be regulated, for example by allosteric regulation and by phosphorylation. They are therefore able to perform rather more sophisticated signalling roles than water-soluble second messengers.
Attempts have been made to group intracellular signalling proteins according to their function, but you will soon see that there are plenty that have more than one function, making classification into functional groupings difficult. Nevertheless, these descriptions give a flavour of the variety of possible signalling functions. Later in this chapter we shall discuss many examples from these groups.
Relay proteins simply pass the signal on to the next member of the chain.
Messenger proteins carry the signal from one part of the cell to another. For example, activation may cause translocation of the protein from the cytosol to the nucleus.
Amplifier proteins are capable of either activating many downstream signalling proteins or generating large numbers of second messenger molecules; they tend to be enzymes such as adenylyl cyclase, which synthesizes cAMP, or ion channels such as Ca2+ channels, which open to release Ca2+ ions from intracellular stores.
Transducer proteins change the signal into a different form. Voltage-gated Ca2+ channels are examples of signalling proteins, which fall into two of these functional categories, since in addition to their role as an amplifier protein, they detect a change in membrane potential, and transduce it into an increase in the concentration of a second messenger.
Bifurcation proteins branch the signal to different signalling pathways.
Integrator proteins receive two or more signals from different pathways, and integrate their input into a common signalling pathway.
Modulator proteins regulate the activity of a signalling protein.
Other proteins are involved purely in the correct placement of some signalling molecules:
Anchoring proteins tether members of the signalling pathway in particular subcellular locations, such as the plasma membrane or the cytoskeleton, thereby ensuring that the signal is being relayed to the right place.
Adaptor proteins link one signalling protein with the next at the correct time, without signalling themselves.
Scaffold proteins are proteins that bind several signalling proteins, and may also tether them, forming a much more efficient functional complex. Scaffold proteins may therefore share attributes of both anchoring and adaptor proteins.
1.6 Signalling proteins can act as molecular switches
How does a signalling molecule actually convey a signal? With second messengers, it is easy to understand: they are produced or released in large quantities, diffuse to their target, to which they usually bind, bringing about a functional change, after which they are degraded or stored within a subcellular compartment (such as endoplasmic reticulum). With signalling proteins it is less obvious. Protein concentrations cannot fluctuate rapidly, and protein molecules cannot easily move within the cell. The conformation of many proteins is related to their activity, and is subject to regulatory mechanisms.
What are the mechanisms by which proteins can be switched from one conformation to another?
One way of modulating a protein's activity is by allosteric regulation, whereby binding of a small ligand induces a conformational change in the protein. Another way is by addition of a negatively charged phosphate group, either by phosphorylation of an amino acid residue by a protein kinase or by binding of a GTP molecule instead of a GDP (G proteins).
Although allosteric regulation by binding small molecules is a widespread regulatory mechanism for the activity of many proteins, including receptors and structural, motor and signalling proteins, the addition or loss of phosphate groups usually drives most functional changes in the sequence of activation/deactivation steps that form a typical intracellular signalling pathway. In reality, many intracellular signalling proteins act as molecular switches. What often happens is that the proteins can be temporarily modified, converting them from an inactive (non-signalling) form to an active (signalling) form (Figure 2), or vice versa. Usually the upstream signal induces a change in the protein's conformation, which enables it to carry out its downstream signalling function. The reason why such molecules are sometimes referred to as molecular switches is because they are either ‘on’ or ‘off’. These proteins can be grouped according to how they are switched on/off, rather than their subsequent mode of action. As outlined above and in Figure 8, signalling molecular switches mainly belong to two categories.
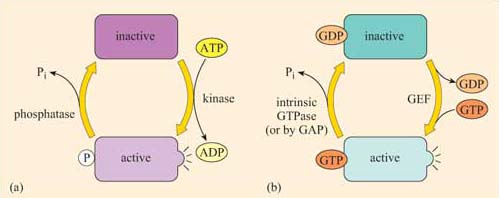
One group of proteins often encountered in signalling are those that are modified by phosphorylation of an amino acid residue by an upstream kinase (Figure 8a). The phosphate is derived from the terminal (γ) phosphate of ATP, and added covalently to a tyrosine, serine or threonine residue by a protein kinase. Phosphorylation usually, but not necessarily, activates a protein. Sometimes, however, it may cause a conformational change that inactivates the protein.
The phosphate group is subsequently removed by a phosphatase, generating Pi (inorganic phosphate), and the protein reverts to its original form. The length of time that the protein remains in its phosphorylated state before being dephosphorylated can be important in determining the signalling outcome. If phosphorylation induces activation, the longer a signalling protein is active, the more downstream signalling molecules it can activate (or the longer that second messengers are synthesized or released by an active signalling protein, the higher the concentrations that they achieve). It is important to note here that many phosphorylated signalling proteins are protein kinases themselves, whose activation results in a series of phosphorylation cascades, as you will see in Section 3.6 (see also Box 1).
The second main group of signalling molecular switch proteins are the GTP-binding proteins, known as G proteins (Figure 8b). In this case, the on/off state characterized by the addition/loss of a phosphate group is not mediated by covalent binding of a phosphate group, but by the binding of a GTP molecule and its hydrolysis to GDP.
In the same way that the rate of dephosphorylation of a phosphorylated protein determines how long it remains active, the length of time that a GTP-binding protein remains active (and hence the number of downstream molecules it can activate) is determined by the rate of GTPase activity. In a sense, GEFs play a similar role to protein kinases and GAPs are comparable to protein phosphatases. In their active form, G proteins also cause a cascade of phosphorylation events, ultimately resulting in a cellular response.
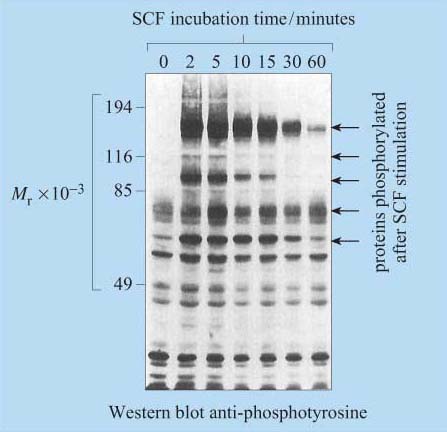
Box 1 Identification of phosphorylated residues in signalling proteins
For many years, phosphopeptide and phosphoamino acid mapping has been a useful method used for identifying protein phosphorylation sites. Cells are metabolically labelled with radioactive Pi, and protein extracts are subjected to polyacrylamide gel electrophoresis (SDS–PAGE and Western-blotted onto a special membrane. The protein of interest is then isolated, hydrolysed into peptide fragments by proteases or into individual amino acids by hydrochloric acid, and are then separated by two-dimensional thin-layer chromatography on cellulose plates. The extent of phosphorylation of tyrosine, threonine and serine residues is finally established by autoradiography. Another technique, first developed in the 1980s, involves the use of monoclonal anti-phosphotyrosine antibodies, which specifically recognize phosphorylated tyrosine residues in many proteins. For investigation of signal transduction mechanisms, this was an essential tool for studying the activity of tyrosine kinases and phosphatases. The antibody can either be used to probe Western-blotted proteins (Figure 9) or, in a more refined technique, can be used to immunoprecipitate the phosphoproteins before separating them by SDS–PAGE.
However, these techniques require the use of populations of single cell types, as these antibodies would not differentiate between cell types in mixed cell populations.
More recently, other polyclonal and monoclonal antibodies targeted to phosphorylated residues (serine, threonine and/or tyrosine) within a specific amino acid sequence of a protein have been developed. For example, there are antibodies that recognize phosphorylated Tyr 527 of Src, and others that recognize Tyr 416 of Src, providing a rapid and easy experimental methodology for the study of Src activation. The use of antibodies specific for phosphorylated amino acid residues has allowed the study of signalling protein activation in vivo on tissue sections using immunocytochemical techniques. Cocktails of 30 or more of these antibodies can also be used in combination to simultaneously detect the activation state of several signalling pathways by probing proteins separated in 2-D gels.
Molecular switches can be a lot more sophisticated than a single on/off function. A protein can be phosphorylated at multiple sites, which may have different effects on its activity.
Integrate many different signals such that the signalling outcome is determined by the summation of signalling inputs. Therefore, they behave as specific signal integrators.
1.7 Localization of signalling proteins
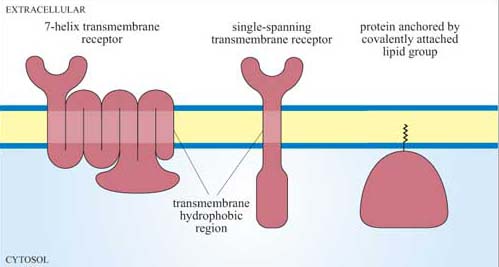
Since signalling proteins cannot diffuse as rapidly as small second messengers, they need be close to their downstream target in order to be able to function. Where they are located with respect to both their subcellular position and their immediate neighbours is therefore vitally important. The plasma membrane is usually the initial location, and proteins can be attached to the plasma membrane in various ways (Figure 10). Many have hydrophobic regions that are inserted into the membrane as the polypeptide is being synthesized (for example, transmembrane receptors).
What post -translational modifications could serve to anchor a signalling protein to the cytosolic side of the plasma membrane?
Covalent addition of a lipid group, prenylation or fatty acylation, tethers proteins to the internal surface of the plasma membrane. The Ras family of G proteins is an example of this type of protein.
The area of the cell membrane near a receptor can become crowded with signalling molecules. Very often, several signalling pathways will need to be activated following binding of the ligand to the membrane receptor, since the cellular response may require multiple changes in cell behaviour (such as a change in cell shape, altered metabolism or changes in gene expression). Many signalling molecules, leading to different signal transduction pathways, will be packed together around the cytoplasmic domain of the receptor, and it is unclear how unwanted signalling outcomes are avoided and how signal specificity is maintained.
One mechanism involves the signalling molecules being arranged on a protein scaffold, such that the proteins are ordered in the correct signalling sequences or, in other words, as a preassembled signalling complex (Figure 11a). This scheme requires one of the signalling components to be able to detach itself from the complex and distribute the signal to other parts of the cell. A similar strategy congregates receptors together with many signalling proteins into specific areas in the plasma membrane such as cholesterol- and glycosphingolipid-rich lipid rafts, which may then be considered as plasma membrane signal initiator units.
Alternatively, complexes can form transiently, following receptor activation. In this case, the intracellular signalling proteins only assemble once the receptor has bound its extracellular signal molecule (ligand). A common mechanism involves the autophosphorylation of key amino acid residues in the cytoplasmic domain of the receptor after ligand binding. The signalling proteins then recognize and dock onto particular phosphorylated amino acids (Figure 11b). We shall see specific examples of this in Section 3.
At some point, the signal has to be transmitted over a significant distance and between cellular compartments within the cell in order to reach its targets. How does the signal ultimately escape from the cytosolic side of the plasma membrane? As pointed out earlier, one mechanism involves the deployment of small, diffusible second messengers, which also results in amplification of the signal throughout the cell. However, this system lacks specificity in the subcellular target. Alternatively, signalling proteins themselves can be directed specifically to another part of the cell. In order to achieve this, their ‘on switch’ (when they are activated by an upstream signalling molecule) must somehow enable them to be transported. For example, when the signalling enzyme MAP kinase is phosphorylated on tyrosine and threonine residues by its upstream kinase, it translocates from the cytoplasm into the nucleus, where it phosphorylates specific transcription factors and so alters the pattern of gene expression. (The MAP kinase pathway is considered in more detail in Section 3.6.)
1.8 Protein–protein interactions in signal transduction
Many signalling proteins have both a catalytic domain and sometimes several binding domains.Some only have binding domains, enabling their proteins to act as adaptor, scaffold or anchoring proteins to bring other proteins together. Because of this multiplicity of binding domains, signalling proteins can potentially combine to form complexes with many other proteins; these complexes may be either transient (e.g. in response to stimulation by a growth factor), or stable (to target a protein to an appropriate location). However, protein–protein interactions are not random, as the specific interactions between binding domains and their recognition sites will determine the precise route(s) that a signal transduction pathway will take.
Figure 12 shows a hypothetical signalling cascade, drawn to illustrate how different protein domains have specific functions that result in an ordered network of consecutive protein–protein interactions – in other words, in a signal transduction pathway. Receptor activation by an extracellular signalling molecule leads to the phosphorylation of tyrosine residues on the receptor and of inositol phospholipids on the cytosolic face of the plasma membrane , thereby creating temporary docking sites for an array of SH2- and PH-containing signalling proteins. A cytosolic signalling protein (shown as signalling protein X) contains three different binding domains plus a catalytic kinase domain. On stimulation by an extracellular signalling molecule, signalling protein X translocates to the plasma membrane by virtue of interactions between its SH2 domain and a phosphorylated tyrosine on the receptor protein (sometimes referred as phosphotyrosine or pY), and between its PH domain and phosphorylated inositol phospholipids in the cytosolic leaflet of the lipid bilayer. This translocation results in a change of conformation in protein X, which unfolds a PTB domain, allowing it to bind a phosphorylated tyrosine in protein Y. The kinase domain in signalling protein X then phosphorylates signalling protein Y on another tyrosine, which subsequently binds to the SH2 domain of an adaptor protein. The SH3 domain in the adaptor protein binds to a proline-rich motif on signalling protein Z. This interaction brings protein Z close to protein Y, such that protein Z is phosphorylated at a tyrosine residue. The signal is then relayed downstream by the activated protein Z.
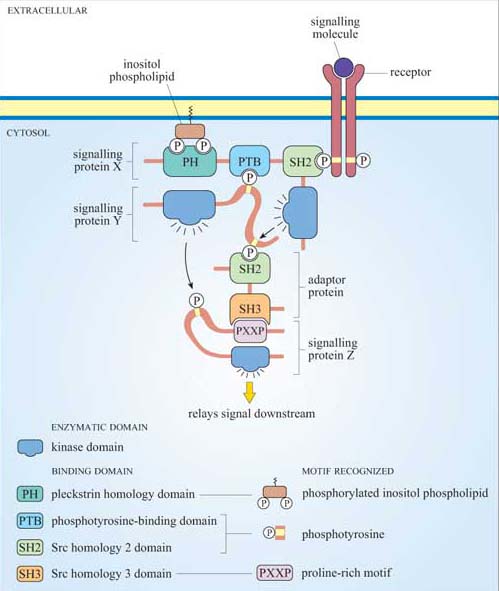
Figure 13 shows the diversity and flexibility of protein-binding domains in some examples of signalling proteins (discussed later in this chapter).
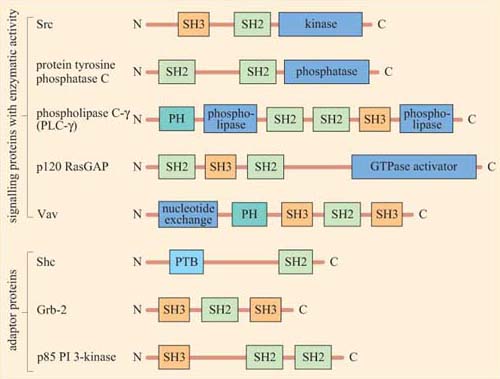
Protein domains can often be identified from their amino acid sequence, and their function deduced from similar, better characterized, proteins. Hence, when a new signalling molecule is identified, it is now often possible to predict, in general terms, from its sequence what it is likely to bind to, and what type of binding domains the signalling molecule contains. It is important to note that whereas the function of a binding domain may sometimes be predicted by the sequence (SH2 domains always bind phosphorylated tyrosines), protein–protein interactions are highly specific — that is, not all phosphorylated tyrosines are recognized by a particular SH2 domain.
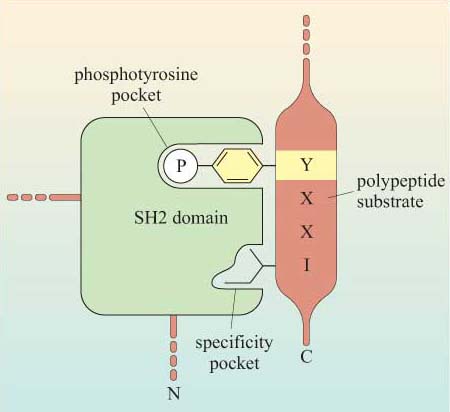
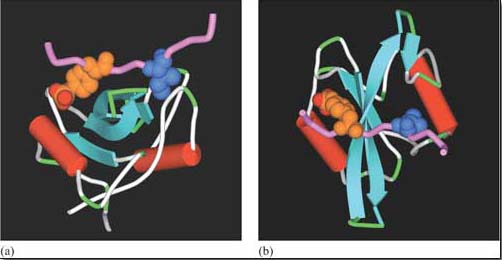
The selectivity of recognition of a motif by a binding domain such as SH2 is conferred by the amino acid sequence adjacent to the phosphorylated residue. We shall illustrate this principle with the SH2 domain of, the tyrosine kinase Src, which has both SH2 and SH3 domains, and a kinase domain (Figure 14a). The core structural elements of its SH2 domain comprise a central hydrophobic antiparallel β sheet, flanked by two short α helices (Figure 14b), which together form a compact flattened hemisphere with two surface pockets. The SH2 domain binds the phosphotyrosine-containing polypeptide substrate via these surface pockets (Figure 15). One pocket (phosphotyrosine pocket) represents the binding site for phosphotyrosine, whereas the specificity pocket allows interaction with residues that are distinct from the phosphotyrosine, in particular the third residue on the C-terminal side of the phosphotyrosine. So, for example, the SH2 domain of Src recognizes the sequence pYXXI, where X is a hydrophilic amino acid, I is isoleucine and pY is phosphorylated tyrosine. Note that all proteins that contain this sequence of amino acids are putative binding partners for the SH2 domain of Src, including the C-terminal phosphotyrosine (pY 527) of Src itself.
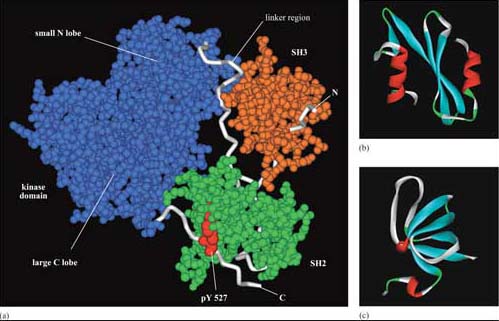
The SH3 domain has a characteristic fold consisting of five β strands, arranged as two tightly packed antiparallel β sheets (Figure 14c). The surface of the SH3 domain bears a flat, hydrophobic ligand-binding pocket, which consists of three shallow grooves defined by aromatic amino acid residues, which determine specificity. In all cases, the region bound by the SH3 domain is proline-rich, and contains the sequence PXXP as a conserved binding motif (where X in this case is any amino acid).
There are various ways of assaying whether signalling proteins interact with each other through their binding domains such as co-immunoprecipitation, yeast two-hybrid screening, proteomics and FRET. Box 2 describes another technique used to analyse protein–protein interactions that you will use in Experimental investigation 3 at the end of this chapter.
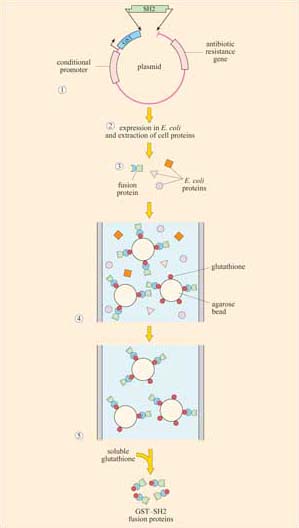
Box 2 Use of fusion proteins for pull-down assays in the study of signalling protein domain interactions
Individual domains often retain their function when isolated from their parent protein, and they can be genetically engineered to be fused with other proteins/peptides. Recombinant fusion proteins consist of two proteins: (a) a protein or peptide sequence used as a tag to facilitate protein isolation; and (b) the ‘bait’ protein, used as a means of indirectly ‘pulling down’ interacting proteins. One very useful example of this technique is the use of glutathione S -transferase (GST) fusion proteins; they consist of GST (tag) fused to a protein or part of a protein of interest (bait).
Using recombinant DNA technology, the DNA encoding the domain of interest is inserted into a plasmid vector just downstream of, and in the same translation reading frame (ORF) as the gene for GST. Under optimized conditions, certain strains of bacteria are induced to take up the plasmid and grown in selection media. Expression of the fusion protein is then chemically induced in transformed bacteria, which are subsequently lysed in a detergent solution. GST is used as the fusion partner because it binds glutathione, a property that can be exploited to purify the fusion protein by affinity chromatography. Free glutathione can be used to elute the fusion protein from the column. The GST can then be cleaved from the protein being investigated, if not further required. This gentle technique produces fusion protein of sufficient quantity and quality for use in, for example, binding assays and enzyme activity assays. Figures 16 and 17 show one example of its use.

1.9 Summary
In a basic model of signal transduction, a signalling molecule binds to a specific receptor, and this activates a sequence (or web) of intracellular signalling molecules that spread the information to relevant parts of the cell, activating target molecules, which effect a cellular response.
Signalling between cells can be contact dependent or via secreted signalling molecules. The latter comprise paracrine, autocrine, endocrine or electrical signalling.
There are four types of cell surface receptors: ion channel receptors, 7-helix transmembrane receptors, receptors with intrinsic enzymatic activity, and enzyme-associated (recruiter) receptors. Receptors with intrinsic transcriptional activity are mostly intracellular.
Two basic categories of signalling molecules intervene in signal transduction, according to the spatial and temporal requirements of the signalling pathway.
a.Small diffusible signalling molecules (‘second messengers’) enable rapid signal amplification and a widespread cellular response.
b.Signalling proteins fulfil many roles (by virtue of protein–protein interaction and protein regulation), including signal integration, modulation, transduction and anchoring functions.
G proteins and proteins activated by phosphorylation on tyrosine, serine and/or threonine residues can act as molecular switches.
The subcellular location of the signalling protein is critical to its function, and this is aided by transient or preassembled signalling complexes.
Specific binding of signalling proteins to each other is critical for the effective transduction of the signal. Binding domains allow transient binding to specific (often phosphorylated) amino acid sequences or to phospholipids.
2 Receptors and their ligands
2.1 Introduction
Every receptor has to be able to recognize its particular ligand in a specific manner, and become activated by it in such a way that it transmits the signal to the cell. We shall deal with receptor specificity and activation mechanisms. Then we shall see how the same principles of specificity and activation also apply to intracellular receptors.
2.2 Receptor specificity
Binding of an extracellular signal to its receptor involves the same type of interactions as those between an enzyme and its substrate. Receptor specificity depends on the binding affinity between the ligand and the binding site on the receptor. The dissociation constant (KD) describes the affinity between receptors and their ligands.
Proteins can be thought of as consisting of various domains, and the different combinations of structural motifs in the extracellular regions of receptors will confer the specificity of a receptor for its ligand. Ligand binding may involve multiple sites of contact between the ligand and different domains of the receptor. It is possible that some interactions between the ligand and its receptor may be important for binding, whereas others may be necessary for signal transfer. An example of a ligand that binds to a 7TM receptor is C5a, a chemoattractant cytokine (called a ‘chemokine’). The interaction is an association of the C5a N-terminus with a pocket within the receptor, involving its extracellular loops 2 and 3, and the N-terminus. This interaction is not in itself sufficient for receptor activation. For this to occur, the C-terminus of C5a must bind to other sites in the bundle formed by the receptor's seven α-helical membrane-spanning segments.
Ligands are classified as either receptor agonists or antagonists, depending on the outcome of interactions between ligand and receptor. Agonists usually work by binding to the ligand binding site and promoting its active conformation. Antagonists bind to the receptor, but do not promote the switch to the active conformation. In addition, it is not always the case that one ligand binds specifically and uniquely to one particular receptor. A single receptor may be able to bind several different ligands, and a single ligand may be able to bind to several receptors.
Here we shall describe two clinically important and well-known examples of receptor–ligand interactions – acetylcholine, a ligand for two structurally different classes of receptors, and adrenalin, which, together with noradrenalin, binds to a number of closely related receptors. (Adrenaline and noradrenaline are also known as epinephrine and norepinephrine, respectively.) We shall return to these receptor signalling pathways throughout the rest of the chapter to illustrate further general principles of signal transduction.
Acetylcholine (ACh) is a neurotransmitter that is released from neuron presynaptic terminals. At the neuromuscular junction, neuron terminals contact a specialized region of skeletal muscle (called the ‘motor end-plate’), where acetylcholine functions as a primary neurotransmitter by stimulating muscle contraction (see Figure 7). In addition, acetylcholine also acts as a neurotransmitter in the heart, where its release slows down the contraction rate. However, ACh receptors (sometimes collectively termed ‘cholinergic receptors’) are structurally and functionally distinct in these two different tissues, and consequently have completely different sets of agonists and antagonists (in both tissues, ACh receptors normally bind to acetylcholine, their endogenous ligand; Figure 18).
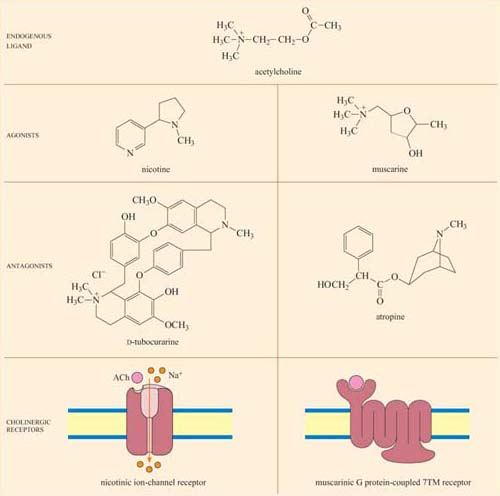
In skeletal muscle, the ACh receptors are ion-channel receptors (Figure 18 and Section 2.3), and are also known as nicotinic receptors. The skeletal muscle subtype of the nicotinic receptor can bind two naturally occurring powerful toxins, the polypeptide α -bungarotoxin (found in the venom of the krait snake) and the alkaloid tubocurarine (found in curare, a poison extracted from the bark of certain trees in South America, and used in the arrows of some tribes). These toxins act as antagonists, and bind reversibly, but with higher affinity to the nicotinic receptor than acetylcholine itself. For example, the KD of α -bungarotoxin is 10−12 to 10−9 mol 1−1, whereas acetylcholine binds with relatively moderate affinity to nicotinic receptors; its KD is 10−7 mol 1−1. As a result, they prevent the action of ACh by binding to and blocking its receptor without activating it, resulting in paralysis and eventually death.
Cardiac muscle contains the other type of ACh receptor, which is a G protein-coupled 7TM receptor (GPCR). This, and the several related subtypes expressed in other neural locations, are usually called muscarinic receptors. Atropine is a naturally occurring antagonist of muscarinic receptors, and is derived from the berries of deadly nightshade, Atropa belladonna (so called because extracts of it were applied to the eyes by women in the Renaissance period, which resulted in a ‘doe-eyed’ beauty). One of the downstream consequences of binding of acetylcholine to muscarinic receptors in cardiac muscle cells is the opening of K+ion channels, which causes membrane hyperpolarization and a decrease in the heart's contraction rate.
Because the muscarinic and nicotinic receptors are not structurally related, there is no overlap between their major agonists and antagonists; nicotine, α-bungarotoxin and tubocurarine have no effect on muscarinic receptors, whereas muscarine and atropine have no effect on nicotinic receptors. How, then, can acetylcholine be a common agonist and bind to two completely different groups of receptors, if their other agonists and antagonists are restricted to binding to just one type each? The answer lies in the flexibility of the acetylcholine molecule. Most of the agonists and antagonists have relatively rigid ring structures, whereas acetylcholine is able to adopt different conformations (Figure 18, top), which may help it adjust to the different binding sites in the two receptors.
Adrenalin is the classic ‘fight or flight’ hormone, having effects on multiple tissues that help put together an appropriate coordinated response to a situation of danger. Actions as varied as increased heart rate, dilation of relevant blood vessels (especially those in skeletal muscle) and constriction of other blood vessels (especially in the skin and digestive tract), and mobilization of metabolic fuels (such as glycogen in liver and skeletal muscle, and stored fat in adipose tissue) are all part of this response.
These effects are mediated by two classes of structurally related GPCRs, the α and β adrenergic receptors, which have the subtypes α1 and α2, and β1, β2 and β3, each with different tissue distributions (Figure 19). Figure 20 shows the range of effects of α and β adrenergic receptors in various tissues and organs.
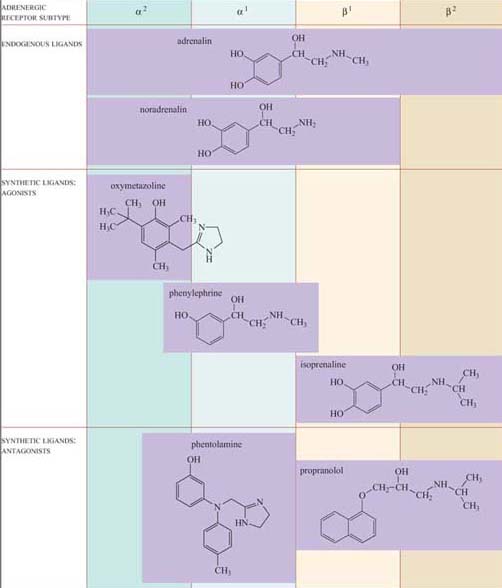
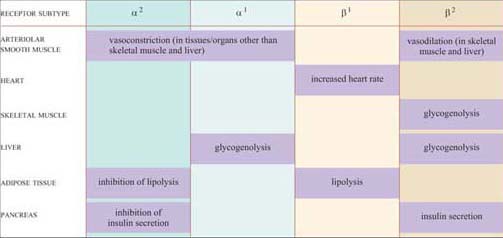
For example, the smooth muscle surrounding arteries in the digestive tract contains predominantly α receptors, which mediate vasoconstriction, and the smooth muscle surrounding arteries in skeletal muscle contain predominantly β receptors, which mediate vasodilation. Note also that the β2 adrenergic receptor increases insulin secretion by pancreatic cells, whereas the α2 adrenergic receptor reduces it.
The classification of adrenergic receptors is based on their interactions with various synthetic agonists and antagonists, which probably reflects the receptor structures. In the simplest example, both α receptors are stimulated by the agonist phenylephrine, and β receptors are exclusively stimulated by the agonist isoprenaline. On the other hand, phentolamine acts as an antagonist of α receptors, whereas propanolol acts as an antagonist of β receptors. Adrenalin itself, of course, can bind all the receptor subtypes, but the structurally related noradrenalin (which is also released into the circulation), has a more restricted binding profile and much more limited effects.
2.3 Receptor activation
Receptors may be activated by conformational change (for example, ion-channel receptors such as nicotinic receptors, and 7TM receptors such as muscarinic receptors and adrenergic receptors), by formation of dimers (such as receptors with intrinsic enzymatic activity and recruiter receptors) or by proteolysis. We shall now consider how each cell surface receptor class described in Section 1.3 is activated.
2.3.1 Ion-channel receptors
Nicotinic cholinergic receptors are probably the best studied of all receptors, firstly because they are present throughout skeletal muscle, and secondly because there are plenty of natural and synthetic toxins that bind specifically to this receptor. Furthermore, the technique of patch-clamp electrophysiology has made possible the detailed characterization of the properties of individual ion channels (Figure 21a).
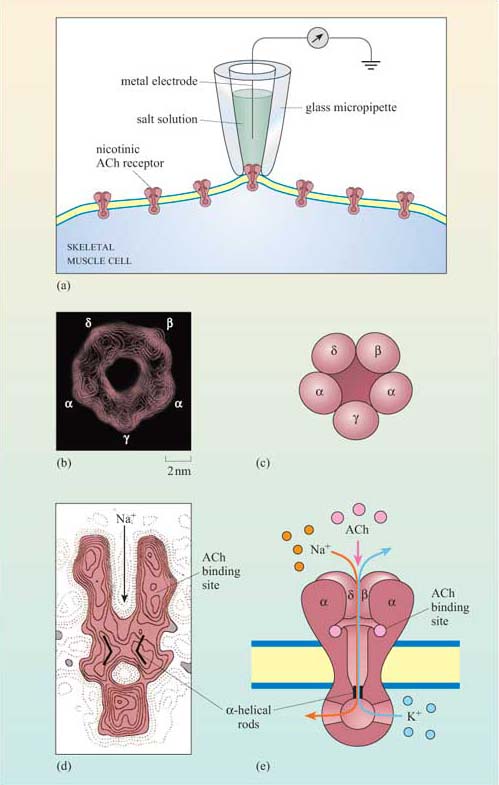
Nicotinic receptors are composed of five subunits (two α subunits together with one each of the β, γ and δ subunits), which assemble to form a pore in the membrane (Figure 21b–e). The pore can switch between an open and a closed state on binding of two molecules of acetylcholine to the two α subunits at sites within the channel (Figure 21). Although the channel alternates between an open and a closed state, binding of acetylcholine increases the probability of the channel being in its open state. When the channel is open, sodium ions flow into the muscle cell, using concentration and voltage gradients. The influx of positive charge due to the Na+ions inside the cell tends to locally neutralize the negative charge inside the cell (called ‘depolarization’). The channel is also permeable to K+ions, which exit the cell. However, the overall effect of the movement of ions causes the net charge inside the cell with respect to the outside to become more positive, and this is ultimately responsible for skeletal muscle contraction (Figure 7).
2.3.2 Seven-helix transmembrane (7TM) receptors
Although in unicellular organisms such as the yeast S. cerevisiae there are only two classes of 7TM receptors, the pheromone and glucose receptors, multicellular organisms have many more, accounting for up to 5% of all genes in C. elegans and 2% of genes in the human and Drosophila genomes. 7TM proteins have been classified into four classes, A, B, C (Table 1). Between them, they can bind a huge range of ligands including simple ions, nucleotides, lipids, steroids, modified amino acids, peptides and glycoprotein hormones at a variety of binding sites, and even photons can activate certain 7TM receptors. An example of the binding of a ligand (adrenalin) to its 7TM receptor is shown in Figure 22. As a result of this wide range of ligands, the mechanisms of activation of 7TM receptors are also extremely diverse. They may include, for example, proteolytic cleavage of the N-terminus, and subsequent binding of the new peptide fragment to the central core or ligand binding to the N-terminus or to the central core. However, in all cases the end result is a change in the conformation of the 7TM receptor (Table 1).
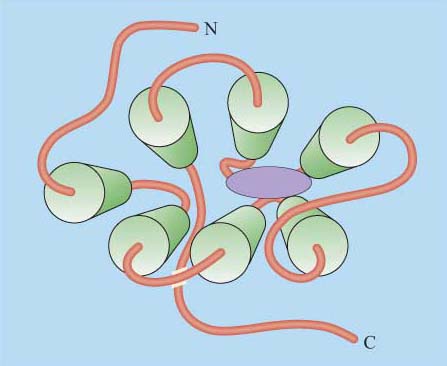
Can you recall any examples of 7TM receptors?
Muscarinic cholinergic receptors and adrenergic receptors.
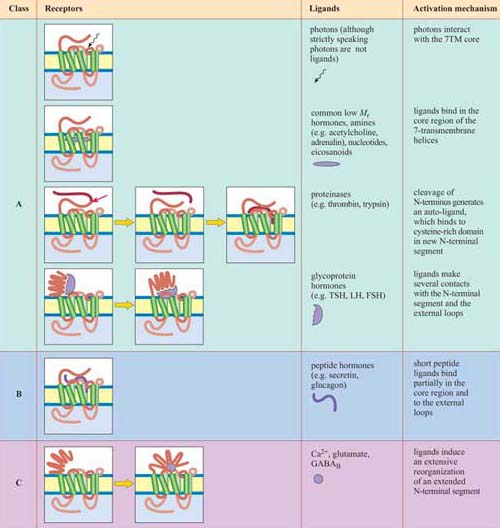
*Activation of the A, B and C classes of 7TM receptors involves coupling to G proteins
In the case of GPCRs, ligand binding influences the equilibrium between the active and inactive conformation of the receptor in favour of the active conformation, altering the interaction of the cytosolic loops of the protein with a trimeric G protein (which may be already associated with the receptor in a preformed complex). In turn, this brings about activation of the G protein (described in Section 3.2).
What is the fundamental difference between signalling through nicotinic and muscarinic ACh receptors?
Nicotinic receptors do not employ a signal transduction pathway to effect their action: the binding of the ligand directly opens a Na+ion channel linked to the receptor, and causes membrane depolarization, resulting in the contraction of skeletal muscle cells. Muscarinic ACh receptors, being GPCRs, activate signal transduction pathways via G proteins.
2.3.3 Receptors with intrinsic enzymatic activity
Receptors with intrinsic enzymatic activity are the second biggest group of receptors after the GPCRs. They include four types according to the form of enzymatic activity of the intracellular domain (Figure 23a).
Receptor tyrosine kinases (RTKs) On activation, the kinase domain phosphorylates tyrosine amino acid residues. There are seven classes of RTK with different extracellular domains (Figure 23b).
Receptor serine–threonine kinases On activation, the kinase domain phosphorylates serine and/or threonine amino acid residues.
Receptor tyrosine phosphatases The intrinsic tyrosine phosphatase activity of the enzymatic domain is suppressed on activation.
Receptor guanylyl cyclases The enzymatic domain generates the second messenger cGMP from GTP following activation.
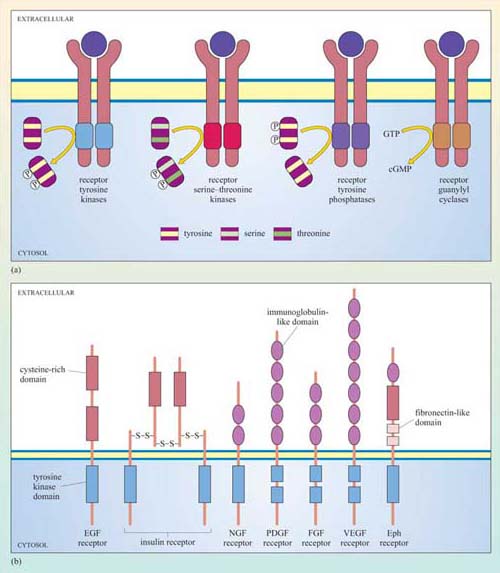
The basic model of activation for receptors with intrinsic enzymatic activity is that ligand binding induces dimerization (in some cases oligomerization) of the receptor, which brings together the cytoplasmic enzymatic domains and leads to a change in enzymatic activity. Dimerization may occur between different receptors that bind the same ligand (heterodimerization), or between the same type of receptor chains (homodimerization), or either. RTKs, RTPs and guanylyl cyclase receptors generally form homodimers (an exception being the epidermal growth factor (EGF) receptor tyrosine kinase), whereas receptor serine–threonine kinases generally form heterodimers. In some cases, oligomerization of several receptors is required for activation.
We shall now describe the general mechanism of activation of RTKs in more detail. There are several strategies by which an extracellular signal may achieve RTK dimerization leading to activation of the receptor:
Ligands such as EGF, which is a monomer, have two binding sites for each receptor unit.
Platelet-derived growth factor (PDGF) is a covalently linked dimer, in which one subunit binds to one PDGF receptor chain, and the other subunit binds to another PDGF receptor chain (Figure 24).
Fibroblast growth factor (FGF) binds to proteoglycans (located on the cell surface or on the extracellular matrix) and induces clustering of FGF receptors.
Ephrins are bound to the plasma membrane of the signalling cell in clusters, and thereby induce association of their receptors (called Eph receptors) on the target cells following cell–cell contact.
The insulin receptor is a tetramer prior to binding insulin: on insulin binding, activation occurs by rearrangement of the different receptor chains that brings the kinase domains in close proximity.
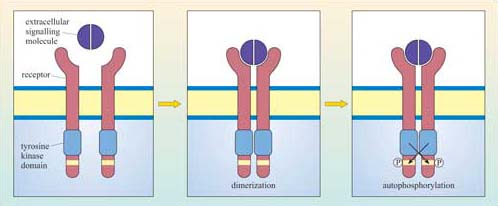
Although there can be a great deal of variation in the extracellular domains of RTKs (Figure 23b) and in the ways the extracellular signal binds to its receptor, the basic mechanism of receptor activation still applies (Figure 24). Association between receptors results in cross-phosphorylation of the kinase domain on each intracellular tail of the RTK, a process called autophosphorylation. This results in an increase in its intrinsic kinase activity, which causes phosphorylation of tyrosines in other parts of the cytoplasmic domain (and/or other proteins). Autophosphorylation generates docking sites on the receptor for downstream signalling proteins that contain SH2 domains.
Many proteins can bind to phosphotyrosine (pY) residues, but these interactions are influenced by nearby amino acid side-chains (see previous section). For example, the PDGF receptor has specific phosphotyrosine sites, which can bind the regulatory (p85) subunit of phosphatidylinositol 3-kinase (PI 3-kinase), a GTPase-activating protein (p120 RasGAP) and phospholipase C-g (PLC-γ), among others (Figure 25). The insulin receptor extends its docking potential by associating with a large protein, insulin receptor substrate 1 (IRS-1), which has many tyrosine residues that can be phosphorylated by the insulin receptor (Section 4). These proteins are called ‘docking proteins’ and may be activated by being directly phosphorylated by the RTK, or by interactions with other docking proteins or plasma membrane molecules. Some docking proteins are adaptor proteins that merely serve to bring other signalling molecules into place. The overall effect of this system is the recruitment of many different signalling pathways, allowing the modulation of many cellular processes.
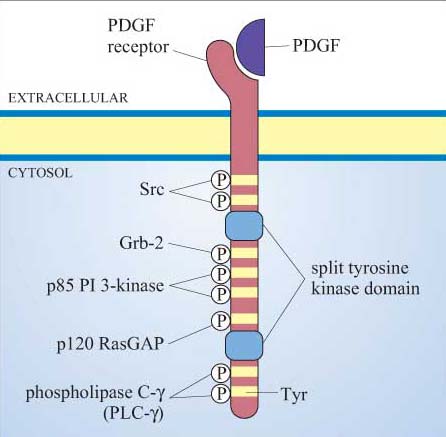
2.3.4 Recruiter receptors
Enzyme-associated or recruiter receptors also form dimers (or oligomers) on activation by their ligand, in a similar way to receptors with intrinsic enzymatic activity. Dimerization facilitates an interaction between the cell surface receptor (which lacks a catalytic domain) and cytosolic proteins with enzymatic activity. In the case of receptors that associate with tyrosine kinases (called ‘tyrosine kinaseassociated receptors’, the most common in this group), it is the non-covalently linked cytoplasmic tyrosine kinase which is autophosphorylated following receptor dimerization. Sometimes homo- or heterodimerization is not sufficient for receptor activation, and activation may follow oligomerization (clustering of several receptors on the membrane) or require membrane-bound co-receptors (structurally unrelated receptors necessary for signal transfer), which may even be RTKs. The end result of the multiplicity of activation combinations for these receptors is that it allows a refinement of signal specificity and diversity, as different downstream effectors are recruited depending on the ligand–receptor complex.
The Src family of tyrosine kinases are the biggest group of kinases that are recruited by tyrosine kinase-associated receptors. One example is Lck (Figure 26). In immune reactions, lymphocyte activation brings together T cell receptors (Figure 5) and other receptors (called ‘CD4’ or ‘CD8’ depending on the activated lymphocyte), which are associated with Lck. Clustering of receptors on the cell surface then results in the tyrosine phosphorylation of the T cell receptor by Lck and activation of downstream signalling pathways. Lck is also very adaptable. As well as associating with CD4 and CD8 receptors, it can also be recruited by means of its SH2 domains to other activated tyrosine kinase (or associated) receptors, thereby strengthening and propagating the signal.
2.4 Receptor inactivation
As with all signalling components, receptors need to be switched off as well as on. Receptor inactivation can operate in several ways including removal of the ligand by degradation or sequestration, and desensitization of the target cell.
Binding of a ligand to its receptor is a reversible process, as the ligand will ultimately dissociate from the receptor and may be degraded. Acetylcholine is a good example of a signal regulated in this way; it is degraded by the enzyme cholinesterase within milliseconds of its release from neuron terminals.
Ligand removal may also occur by sequestration following binding to proteins other than its normal receptor (these may be decoy receptors or extracellular proteins). ‘Decoy receptors’ are cell surface receptors that bind the ligand but do not convey the signal onward in the pathway (for example, truncated RTKs that lack the intracellular kinase domain). Similarly, soluble extracellular proteins containing ligand-binding domains may also sequester the ligand. In both cases, the effect of the extracellular signal is neutralized prior to receptor binding.
If the ligand cannot be degraded or sequestered, the target cell may, after prolonged activation, become desensitized. Desensitization can occur in several ways, the principal ones being inactivation of the receptor (blocking its interaction with downstream signalling components), sequestering the receptor into endocytic vesicles (from which it can be recycled back onto the plasma membrane), or ultimately degrading the receptor in lysosomes. These mechanisms of receptor desensitization usually function in sequence, and progression from one stage to another can depend on factors such as ligand concentration. Activated GPCRs can be desensitized when they are phosphorylated by different protein kinases. The phosphorylated receptor then binds to a cytosolic protein called arrestin, forming a complex that both blocks any interaction with downstream signalling molecules and couples the receptor to clathrin-coated pits , inducing receptor-mediated endocytosis. Another example of desensitization induced by binding of a cytosolic protein to the receptor is provided by c-Cbl. It binds to phosphotyrosine residues of certain activated RTKs via its SH2 domains, thereby promoting the association of the receptor–Cbl complex with ubiquitin. The receptors are then sequestered and degraded via the ubiquitin–proteasome pathway.
2.5 Intracellular receptors
Signal receptors are usually located at the cell surface. However, it is important to remember that there are some groups of receptors that do not fit into the general signal transduction model set out in Figure 2, These are intracellular receptors, which bind small or lipophilic molecules such as steroid hormones, which can cross the cell membrane. The signalling pathways activated by these receptors seem quite simple compared with the other pathways we shall be dealing with, but the same principles of ligand binding, conformational change, signal amplification, translocation and so on described earlier still apply.
One important family of intracellular receptors are the nuclear receptors (also known as ‘nuclear hormone receptors’), which includes receptors for steroid hormones, thyroid hormones, retinoids and vitamin D. Although the ligands differ in their structural type, all nuclear receptors are structurally similar. They are good examples of receptors with intrinsic transcriptional activity (Section 1.3), comprising a transcription-activating domain, a DNA-binding domain and a ligand-binding domain. Their ligands are all small and hydrophobic, and so they can diffuse readily through the plasma membrane. The receptors are usually held in an inactive conformation by inhibitory proteins (often chaperones/heat-shock proteins). Binding of the ligand induces a conformational change that causes the inhibitory protein to dissociate from the receptor (Figure 27). The receptor may then translocate to the nucleus if it was in the cytoplasm, or it may already be in the nucleus; either way, the receptor–ligand complex is now able to bind to specific DNA sequences by means of its DNA-binding domain. Binding to DNA can also be facilitated by association of the receptor–ligand complex with other proteins (referred to as ‘coactivator proteins’). The DNA sequence to which the receptor–ligand complex binds is a promoter region of the target genes; in the case of hormones, it is called a ‘hormone response element (HRE)’.
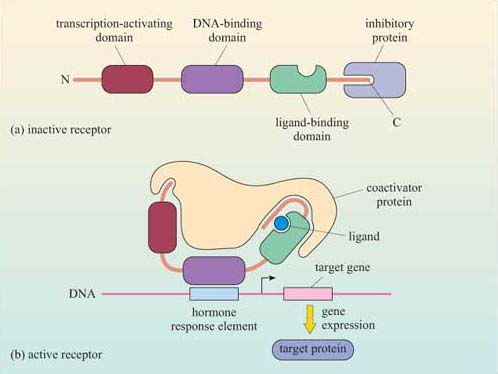
2.6 Summary
Receptors comprise a limited number of structural motifs, which determine binding affinity and specificity of receptor–ligand complexes. Some ligands bind to several receptors and some receptors bind to several ligands.
Acetylcholine is a good example of a ligand with two structurally different kinds of receptor. Nicotinic receptors are ion channels, which are found predominantly in skeletal muscle, and are stimulated by nicotine. Nicotinic receptor antagonists include the toxins α -bungarotoxin and tubocurarine. Acetylcholine binds at two sites within the channel. Muscarinic receptors, in contrast, are 7TM G protein-coupled receptors, found (for example) in cardiac muscle. Muscarine acts as an agonist, whereas atropine acts as an antagonist. Acetylcholine binds in the core region (Table 1) of the transmembrane helical segments.
Adrenalin, however, has a range of structurally related 7TM G protein-coupled receptors, with different tissue distributions and different affinities for numerous agonists and antagonists. Adrenalin, like acetylcholine, binds in the core region of the receptor, though other GPCRs can be activated in a variety of ways.
Mechanisms for receptor activation are varied and include conformational changes (ion-channel receptors and 7TM), homo- or heterodimerization (receptors with intrinsic enzymatic activity and recruiter receptors) or even proteolysis.
For most 7TM receptors (the exception being the Frizzled class of 7TM receptors), conformational change on ligand binding activates associated cytoplasmic G proteins. Hence, they are called ‘G protein-coupled receptors’.
Receptors with intrinsic enzymatic activity include receptor tyrosine kinases, receptor serine–threonine kinases, receptor tyrosine phosphatases, and receptor guanylyl cyclases. Most RTKs are activated by dimerization on ligand binding, leading to autophosphorylation of the cytoplasmic portion of the receptor. Phosphorylated tyrosine residues serve as docking sites for SH2-containing signalling proteins, which also recognize sequence-specific flanking motifs.
Dimerization of recruiter receptors facilitates the interaction between the membrane-bound receptor and cytosolic proteins with intrinsic enzymatic activity such as kinases.
Receptors can be inactivated by removal of the ligand, or by receptor desensitization, which can be by inactivation, by sequestration or by degradation of the receptor.
Some signalling molecules can diffuse across the plasma membrane, and so have intracellular, rather than cell surface receptors. Small hydrophobic ligands such as steroid hormones bind to members of the nuclear receptor group, which undergo conformational change and bind to specific DNA sequences, stimulating transcription of target genes.
3 Intracellular signalling components
3.1 Introduction
We are now ready to describe in detail the major intracellular signalling pathways responsible for relaying the signal from the surface receptor to evoke a cellular response. This section will deal with signalling molecules that operate at the cytosolic leaflet of the plasma membrane (trimeric G proteins, monomeric G proteins and lipid-modifying enzymes), second messengers (such as Ca2+, cAMP, cGMP), protein kinases and phosphatases, and finally transcription factors.
3.2 Trimeric G proteins
G proteins are attached to the cytosolic face of the plasma membrane, where they serve as relay proteins between the receptors and their target signalling proteins.
Trimeric G proteins interact with 7TM receptors and are all heterotrimeric, having structurally different α, β and γ subunits. Monomeric G proteins are the small G proteins, such as Ras, which are structurally related to the α subunit of trimeric G proteins.
The three-dimensional structure of trimeric G proteins in their inactive form is shown in Figure 28.
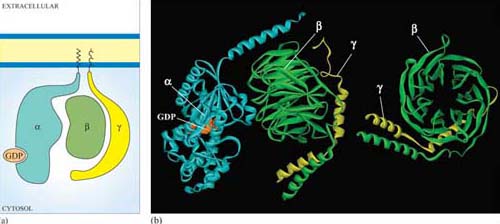
Ligand binding induces a conformational change in the 7TM receptor, which results in the release of GDP and binding of GTP to the α subunit (Figure 29). As a result, the α subunit also changes conformation and becomes activated. This conformational change results in the dissociation of the α subunit from the βγ complex, which also becomes activated, although it does not change conformation itself. The α subunit primarily, and also the βγ complex to a lesser extent, regulate the activity of downstream effector proteins located on the plasma membrane. There are many different α subunits, which can be classified according to sequence similarity, and to which upstream and downstream proteins they interact with (see Table 2 for the most important ones). In fact, the G protein complex is often categorized by the type of α subunit it is formed from; hence you will come across Gαs, Gαi, Gαq, etc. For example, Gαs stimulates adenylyl cyclase, whereas Gαi inhibits it, and Gαq activates PLC-β (see Table 2). There are also different βγ subunits, some of which have been shown to have their own effector function. More generally, though, βγ subunits are thought to stabilize the inactive state of the α subunit.
| Target effector protein | G protein subunit type | Interfering toxin† |
|---|---|---|
| ion channels | regulated by Gαs, Gαi, Gα0 and βγ (for example, Gαi and Gα0 coupled to muscarinic ACh receptor activates K+channels) | |
| adenylyl cyclase | activated by Gαs | cholera toxin |
| inhibited by Gαi | pertussis toxin | |
| cGMP phosphodiesterase | activated by Gαt (transducin) in Photoreceptors | |
| phospholipase C-β | activated by Gαq and Gα0 | |
| phospholipaseA2 | activated by a βγ ? complex | |
| PI 3-kinase | activated by a βγ ? complex | |
| small GTPases | Gα12/13 |
Footnotes
†Cholera toxin and pertussis toxin (from the Bordetella pertussis bacterium, which causes whooping cough) both interfere with the action of G protein α subunits. Cholera toxin locks Gα subunits into an active form and pertussis toxin interferes with Gαi subunits by inhibiting them, making these toxins useful laboratory tools for determining which signalling pathways are activated by GPCRs.G proteins usually remain active for only a short time, which depends mainly on the rate of hydrolysis of GTP to GDP (Figure 29). The intrinsic GTPase activity of the a subunit is quite inefficient by itself. For many cell signalling processes where a rapid turnover rate is necessary (for example, transduction of a photoreceptor activated by visual stimuli), the intrinsic GTPase activity of the α subunit is usually aided by binding of a second protein that enhances the rate of G protein inactivation. This may be either its target protein, ensuring that the α subunit remains active for just as long as it takes to make contact with the target, or a GTPase activating protein (GAP, Section 1.6).
3.3 Lipid-modifying enzymes
The internal surface of the plasma membrane provides a useful environment for spreading signals received by surface receptors around the cell. Several specialist enzymes are activated by membrane-bound receptors, creating large numbers of small lipid-soluble second messenger molecules, which can diffuse easily through the membrane. These enzymes all use phosphatidylinositol (PI) and its derivatives as their substrates. PI itself is a derivative of glycerol: the OH group on carbon atom 1 has been replaced with an inositol ring linked via a phosphate group, and the OH groups on carbon atoms 2 and 3 have been replaced by two fatty acyl chains, one saturated and one unsaturated (Figure 30a).
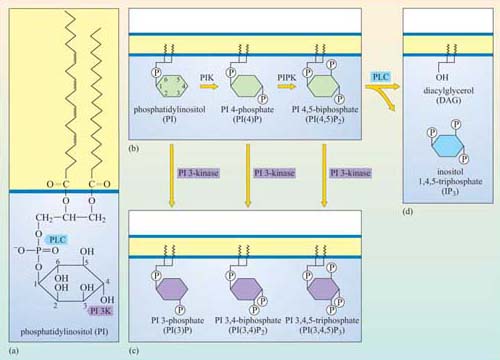
The fatty acyl chains are embedded in the cytosolic leaflet of the plasma membrane, leaving the inositol ring projecting into the cytosol. Carbon atoms 3, 4 and 5 of the inositol ring can be phosphorylated by lipid kinases (Figure 30). The best-studied enzymes employing these substrates are the phospholipase C family, which cleave the fatty acyl chains from the inositol ring, and phosphatidylinositol 3-kinase (PI 3-kinase), which phosphorylates carbon atom 3 of the inositol ring. These products then serve as second messengers. We shall now briefly explain the action of these enzymes, and then go on to describe the roles of the second messengers they generate.
3.3.1 Phosphatidylinositol 3-kinase (PI 3-kinase)
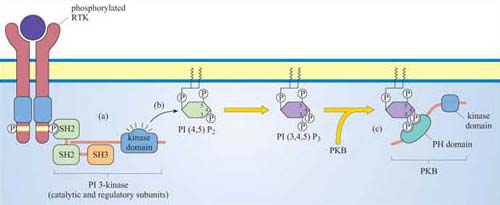
Members of this family of lipid kinases usually have two subunits: one is a catalytic subunit with a lipid kinase domain and the other is a regulatory subunit, which contains two SH2 domains and a SH3 domain (p 85 PI 3-kinase in Figure 13).
What will the SH2 domains of the regulatory subunit enable PI 3-kinase to do?
They will enable PI 3-kinase to bind to proteins containing a phosphorylated tyrosine residue within a specific motif. In this way, PI 3-kinase is targeted to the membrane when required, by binding to phosphotyrosine residues on activated RTKs.
The preferred substrate of PI 3-kinase is PI(4,5)P2. However, this kinase also phosphorylates PI and PI(4)P. It is the phosphorylation at the 3 position by PI 3-kinase that makes the molecule active in a signalling context. Thus, the two main products, phosphatidylinositol 3,4-biphosphate (PI(3,4)P2) and phosphatidylinositol 3,4,5-triphosphate (PI(3,4,5)P3), are both active signalling molecules because they are recognized by PH domains in other proteins (Figure 31). In contrast, PI(3)P is not an active second messenger.
What distinguishes PI(3,4)P2, PI(3,4,5)P3 and PI(4,5)P2 from each other? Which are substrates for PI 3-kinase?
These molecules differ in their phosphorylation state. PI(3,4) P2 is phosphorylated on carbons 3 and 4, PI(3,4,5)P3 on carbons 3, 4 and 5, and PI(4,5)P2 on carbons 4 and 5. Only PI(4,5)P2 is a substrate for PI 3-kinase, whereas the two others are products.
How does the activity of PI 3-kinase influence the localization of signalling proteins?
By virtue of its catalytic activity, PI 3-kinase generates PI(3,4)P2 and PI(3,4,5)P3, which serve as plasma membrane docking sites for PH-containing proteins.
Proteins differ in their affinity for binding to either PI(3,4)P2 or PI(3,4,5)P3, depending on the interacting PH domain. One signalling enzyme that utilizes the membrane docking sites generated by PI 3-kinase is protein kinase B (PKB, also known as Akt), which thereby becomes an accessible substrate for an upstream kinase, PDK1 (not shown in Figure 31). PKB is a serine–threonine kinase, principally involved in mediating survival signals. Another important target is phospholipase C (PLC), which binds to PI 3-kinase substrates at the membrane via its PH domain (see below). The docking sites for PH domains are, as with all signalling components, temporary; specific inositol phospholipid phosphatases ultimately remove the phosphate from the 3 position of the inositol ring.
3.3.2 Phospholipase C (PLC)
Members of this family of enzymes contain two catalytic domains and several protein binding domains (Figure 13). The PH domain can temporarily tether phospholipase C to the membrane by attachment mainly to PI(3,4)P2.
We shall discuss two main isoforms of PLC: PLC-β, which is activated by a subset of trimeric G proteins (Gαq and Gα0), and PLC-γ, which, in contrast, associates with phosphotyrosines on activated RTKs (such as the PDGF and insulin receptors) by means of its SH2 domains. The substrate of both PLC-γ and PLC-β is PI(4,5)P2, which is cleaved by PLC to produce two second messengers: 1,2-diacylglycerol (DAG) consists of linked fatty acyl chains, and so remains in the plasma membrane; inositol 1,4,5-triphosphate (IP3) consists of the phosphorylated inositol ring, which because it is water-soluble is able to diffuse through the cytosol.
Why is IP3 released from the plasma membrane? You may want to look back at the structure of IP3 (shown in Figure 30).
IP3 is hydrophilic and lacks the hydrophobic fatty acyl chains that anchor inositol phospholipids in the plasma membrane.
IP3 binds to IP3-gated calcium channels on the ER membrane, causing Ca2+ stored in the ER to flood into the cytosol. This activates many proteins, but most notably (in this scenario) the protein kinase C family (PKC, so called because of their Ca2+ dependence). Binding of calcium causes PKC to translocate to the membrane (Section 3.4). Full activation of PKC is complicated and depends on the isoform involved, but generally DAG binds to, and helps to activate, protein kinase C. Thus, the two products of PLC activity are acting in a coordinated fashion on the same protein (Figure 32).
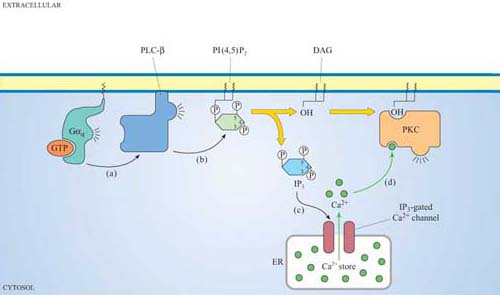
3.4 Second messengers
In the previous section, we have discussed the principles of second messengers (Section 1.5) and, in particular, those produced by PLC (IP3 and DAG) and PI3 kinase (PI(3,4)P2 and PI(3,4,5)P3). We shall now consider the roles and mechanisms of action of the other chief mediators, which are Ca2+ ions, cAMP and cGMP. These are water-soluble second messengers.
Of the second messengers produced by PLC and PI 3-kinase, which ones are hydrophobic?
PI 3-kinase products PI(3,4)P2 and PI(3,4,5)P3 and the PLC product DAG are hydrophobic. They contain fatty acyl chains that anchor them to the plasma membrane.
3.4.1 Calcium ions
The Ca2+ concentration is normally low in the cytosol (~10–7 mol 1–1) compared with the extracellular space (~10–3 mol 1−1). There are several mechanisms for achieving this. The most widespread are ATP-dependent Ca2+ efflux pumps on the plasma membrane, which pump Ca2+ ions out of the cell. Muscle and nerve cells, where oscillations in intracellular Ca2+ concentration often occur, employ an additional Na+-driven Ca2+ exchanger. There are also pumps driving cytosolic Ca2+ into the endoplasmic reticulum, so that it acts as a Ca2+ store. When Ca2+ channels are transiently opened by a signalling protein, Ca2+ floods down the concentration gradient into the cytosol. The result is a rapid 10–20-fold increase in the cytosolic Ca2+ concentration in the cytosol, which in turn activates numerous different Ca2+-dependent proteins, such as PKC.
There are three main types of Ca2+ channel:
a.IP3-gated Ca2+channels on the ER (Section 3.3).
b.Voltage-dependent Ca2+channels in the plasma membrane, which open when the membrane is depolarized. These are used, for example, at neuron terminals, where Ca2+ release stimulates secretion of neurotransmitters.
c.Ryanodine receptors closely associated with receptors on the plasma membrane, which respond to changes in membrane potential, and release Ca2+ from the sarcoplasmic reticulum in skeletal muscle cells (as described in Section 1.5, Figure 7) or from the ER in neurons. Their name derives from their sensitivity to the plant alkaloid ryanodine.
Ca2+-sensitive fluorescent indicators can be used to monitor changes in intracellular Ca2+. After stimulation with an extracellular signal, opening of channels and release of Ca2+ in the cytosol by the mechanisms described above result in local increases in Ca2+ concentration, often circumscribed to small regions of the cell. These increases usually reflect the opening of individual channels or of small groups of channels; these changes in intracellular Ca2+ are called ‘quarks’ or ‘blips’. If the signal is persistent and sufficiently strong, the change in the concentration of Ca2+ spreads across the cell. Under the fluorescent microscope, it appears as if an initial wave of high Ca2+ is followed by other waves propagating through the cytosol, with Ca2+ concentrations first rising and then returning to basal levels; these changes are referred to as ‘spikes’ or ‘oscillations’, and can be repeated at intervals of seconds or minutes. The frequency of Ca2+ oscillations can determine the response. For example, a low frequency of Ca2+ spikes may trigger transcription of one set of genes, whereas a higher frequency triggers transcription of a different set. The sensitivity of the cellular response to the frequency of Ca2+ oscillations requires a special kind of protein called calmodulin.
Calmodulin is abundant, constituting about 1% of total cellular protein. It has no intrinsic catalytic activity, but on binding to Ca2+ it is able to modulate the activity of other proteins. It has four Ca2+ binding sites (small, helical Ca2+-binding motifs called ‘EF hands’, which are also present in some other Ca2+-binding proteins); at least two of them must be occupied for it to adopt its active conformation (Figure 33).
What term describes the regulation of calmodulin by Ca2+?
Calmodulin is allosterically regulated by Ca2+ ions
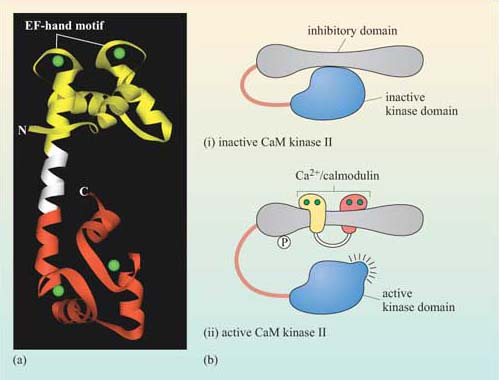
The biggest class of proteins affected by calmodulin is called the Ca2+/calmodulin-dependent protein kinases (CaM kinases), which are serine–threonine kinases. CaM kinase II is found in high quantities in the brain, particularly at synapses. It seems to be involved in some kinds of memory, since mice with mutations in CaM kinase II have specific learning difficulties. On binding Ca2+/calmodulin, CaM kinase II activates itself by autophosphorylation, the degree of activation being dependent on the oscillation frequency of Ca2+ concentration.
Many members of the protein kinase C family are Ca2+-binding proteins, but in these proteins Ca2+ binds to a larger domain, known as the ‘C2 domain’. Ca2+ binding changes the net charge of the C2 domain, enabling it to bind negatively charged phospholipids such as DAG in the plasma membrane (see Section 1.4.2 and Figure 31). Here, Ca2+ is acting as a switch, helping to change the localization of the enzyme.
3.4.2 Cyclic AMP
The concentration of cyclic AMP (cAMP) in the cytosol increases 20-fold within seconds of an appropriate stimulus. This is achieved by the action of the plasma membrane-embedded protein adenylyl cyclase, which synthesizes cAMP from ATP (Figure 34). cAMP is short-lived, as with all second messengers, because it is continuously degraded by cyclic AMP phosphodiesterases to 5′-AMP. Adenylyl cyclase is activated by stimulatory G proteins (Gαs) and inhibited indirectly by inhibitory G proteins (Gαi). It is usually the α subunit of the G protein that regulates adenylyl cyclase in this way, though sometimes the βγ complex can have the same effect. Different hormones can induce a similar cAMP response in a particular cell type, leading to similar cellular outcomes. However, different cell types will respond differently to similar cAMP increases, so the same hormone may have different effects on different cells. For example, cAMP mediates the action of adrenalin acting through β-adrenergic receptors (see Figure 48), causing glycogen breakdown in skeletal muscle cells, while promoting triglyceride breakdown in fat cells. Most of the downstream signals of cAMP are propagated by cAMP-dependent protein kinase A (PKA), which is a serine–threonine kinase.
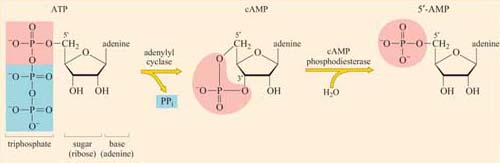
PKA has many target proteins, which vary according to cell type. This explains why a hormone can produce the same increase in cAMP concentration in different cells, yet the cellular response to this increase is different for each cell type. PKA consists of two catalytic subunits and two regulatory subunits. The latter have two roles. They bind to PKA-anchoring proteins, thus tethering the enzyme to particular subcellular locations. They can also each bind two cAMP molecules in a cooperative fashion; when bound to more than two cAMP molecules, the regulatory subunits undergo sufficient conformational change for them to dissociate from the catalytic subunits (which remain in the cytosol). As with other signalling proteins with multiple binding sites, such as calmodulin, PKA is allosterically regulated by the second messenger cAMP. The released catalytic units are now active. They may act in the cytoplasm, for example by phosphorylating enzymes involved in glycogen metabolism and/or they may migrate to the nucleus, where they can switch on transcription of genes containing cAMP response elements (CREs) in their promoters. This is achieved by phosphorylating a serine on the nuclear CRE-binding protein (CREB), which, together with a coactivator, then binds to the CRE and stimulates transcription of the downstream gene.
3.4.3 Cyclic GMP
Cyclic GMP (cGMP) is a second messenger with many similarities to cAMP. It is synthesized from GTP by guanylyl cyclase, and degraded to 5´- GMP by cyclic GMP phosphodiesterases. Some of the targets of cGMP are analogous to those of cAMP: cGMP-dependent protein kinase (PKG), and cGMP-gated Na+ ion channels.
We have already discussed a type of receptor that employs cGMP for signal transfer. What type of receptor is it?
Receptors with intrinsic enzymatic activity with an intracellular guanylyl cyclase catalytic domain (Figure 23a).
3.5 Monomeric G proteins
We shall discuss monomeric G proteins (also called small G proteins or small GTPases) separately from the trimeric G proteins for three reasons: their upstream activators are different, they tend to have different target proteins, and they commonly operate within different signalling pathways.
What structural features and activities do monomeric G proteins share with trimeric G proteins?
You may have thought of the following:
a.Monomeric G proteins are structurally related to the α subunit of trimeric G proteins.
b.They bind GTP and hydrolyse it to GDP, acting as a molecular switch.
c.The rate of GTP hydrolysis can be increased by GAP proteins.
d.They are tethered to the internal surface of the plasma membrane by means of post-translational lipid modifications (Section 1.6).
There are many types of monomeric G proteins, involved in a myriad of cellular processes; examples include Ran, which has a role in the nuclear localization of proteins Rab, with a role in endocytosis or the Rho family, which functions in cell adhesion and migration. The best-studied monomeric G protein is Ras, which we shall consider here in some detail. Ras is structurally similar to the α subunit of trimeric G proteins. In fact, the GTP-binding domain of Gα is known as the Ras domain (Figure 35).
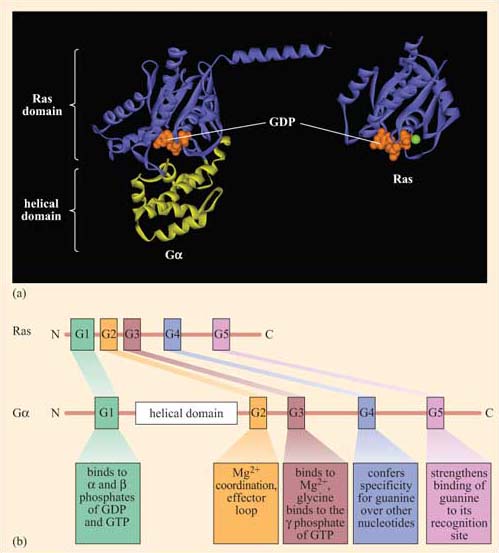
Ras forms part of the signal transduction pathway from most RTKs, including the growth factor receptors. As such, it is central to regulation of the growth, proliferation and differentiation of cells, and is involved in the formation of tumours when these pathways become dysfunctional. Ras homologues have been found to regulate vulval development in C. elegans and photoreceptor development in Drosophila, providing excellent experimental models in which to study its function in development. There are three members of the mammalian Ras family – H-Ras (first discovered in its viral form in Harvey murine sarcoma virus), K-Ras (from Kirsten sarcoma virus) and N-Ras (or neural Ras). There are also more than 70 other small G proteins, which share regions of sequence homology and are involved in a whole variety of cell processes.
For example, Rho and Rac have been shown to mediate changes to the actin cytoskeleton, leading to stress fibre formation and lamellipodia formation, respectively.
Ras is activated following stimulation of many RTKs (Figure 36). How does this happen? It is a much more indirect process than for trimeric G proteins, where conformational change in the receptor directly induces conformational change and activation of the G protein. Instead, what usually happens is firstly that autophosphorylation of RTKs creates docking sites for SH2-containing proteins. One of these SH2-containing proteins is an adaptor protein called Grb-2 (growth factor receptor-bound protein 2). Grb-2 consists of little more than two SH3 domains flanking an SH2 domain (Figure 13). Grb-2 binds to the activated receptor via its SH2 domain. Sometimes the RTK does not contain the correct docking sequences for Grb-2, in which case an intermediary adaptor protein such as Shc (another SH2 protein also in Figure 13) is used.
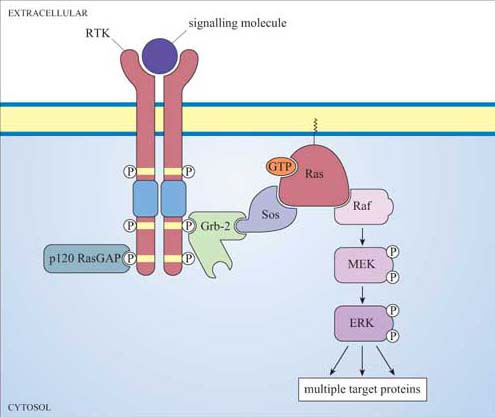
One of the SH3 domains of Grb-2 bind to a Ras activator protein, which contains the appropriate proline-rich domains. Ras activators are proteins that, by binding to Ras, stimulate Ras to exchange its GDP for a molecule of GTP.
What is the name for this class of protein?
Guanine nucleotide exchange factors, GEFs (Section 1.6).
The usual GEF that acts on Ras is called Sos (Figure 36). You can thank Drosophila researchers for this name! The photoreceptor studied by them is called R7, and the gene for the tyrosine kinase receptor involved in photoreceptor development is called ‘sevenless’ (because sevenless mutants lack R7 photoreceptors). The GEF was discovered to be downstream of the receptor, and so its gene was called son of sevenless. Because Sos is recruited to the plasma membrane by Grb-2 following receptor activation, it is then in the right place to bind to Ras (which is permanently tethered there) and encourage it to bind GTP. Since GEFs promote GTP binding and RasGAPs promote hydrolysis of GTP to GDP, the overall model is that GEFs switch Ras on, and GAPs switch Ras off (look back at Figure 8b). GDP-bound Ras is in an inactive conformation, but when it is bound to GTP, two ‘switch’ regions (the main one of which is an α helix) are affected, resulting in the adoption of an active conformation (Figure 37). These switch regions are implicated in binding to effectors and also to RasGAP.
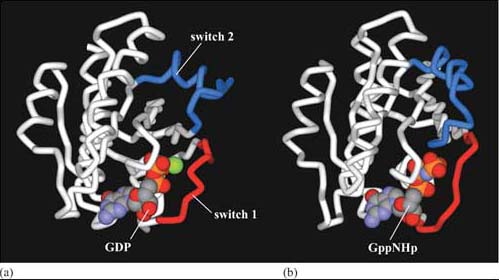
It is remarkable that such a small and permanently membrane-bound protein as Ras is able to mediate so many profound changes within a cell. However, we can see how it can serve as an integrator of signalling information via GEFs and GAPs. As for its effectors, Ras is known to affect PI 3-kinase, but it achieves most of its effects by activating the first component of a key signal transduction cascade called the MAP kinase pathway (Figure 36), which serves to amplify signals and operate over a rather longer time frame than, say, second messenger systems. Moreover, it has many different cellular targets, including transcription factors, which it activates.
3.6 Protein kinases
Protein kinases phosphorylate proteins either at tyrosine residues (tyrosine kinases), or at serine and threonine residues (serine–threonine kinases), or on any of these three amino acids (dual-specificity kinases). All these activities are employed in signal transduction pathways (histidine kinases also operate in certain plant and bacterial pathways, but not in animals). You should now be familiar with receptor tyrosine kinases, and have seen in some detail how phosphorylation of particular tyrosine residues can create docking sites for SH2-containing proteins (Section 2.3). You have also been introduced to the idea that phosphorylating key residues can induce a conformational change that can help to either activate or inactivate the protein, depending on both where the phosphorylation site is on the protein and on what other signals are being received by the protein, as illustrated for Src in Figure 29. You have also become familiar with several of the key serine–threonine kinases involved in signalling, such as the PKA, PKC and CaM kinases (Sections 3.3 and 3.4). There are two further very important groups of kinases that we shall now describe, namely those of the MAP kinase and the JAK–STAT pathways. Then we shall draw all these kinases together into families, and point out common domains and common themes in their mechanism of activation.
3.6.1 The MAP kinase pathway
The MAP kinase pathway is so called because the last component of the pathway was originally identified as a kinase activity in EGF-stimulated cells – hence the name ‘mitogen-activated protein kinase’ (MAP kinase), as it stimulates cell growth and proliferation.
What is the mechanism of activation of the EGF receptor?
The EGF receptor is an RTK (Figure 23b), which becomes activated by dimerization. By contrast with most other RTKs, the EGF receptor does not form homodimers.
It was subsequently found that MAP kinase acts downstream of Ras, and that there are also two intermediary kinases (Raf and MEK in mammals), making it a three-kinase signalling module (Figure 36). MAP kinase forms the link between Ras in the plasma membrane and downstream effectors such as transcription factors in the nucleus. As such, it is crucial to signalling from growth factor receptors and other RTKs (and tyrosine kinase-associated receptors).
When Ras is activated, it recruits the serine–threonine kinase Raf to the membrane, where it becomes activated in a poorly understood mechanism involving membrane-bound kinases other than Ras itself (see Box 3). Raf is the first of the three kinases in the MAP kinase cascade, and is therefore also known as MAP kinase kinase kinase (abbreviated to MAPKKK). Raf phosphorylates (and thereby activates) the next kinase (a MAP kinase kinase, or MAPKK), which in mammalian cells is called MEK (MAP/ERK kinase). In turn, this phosphorylates MAP kinases (the classic one in mammals being ERK, e xtracellular signal r egulated k inase). In their active state, MAP kinases are able to translocate to the nucleus and are able to phosphorylate numerous nuclear target proteins, leading to such major cellular events as proliferation or differentiation.
Why is there a chain of three kinases rather than just one? This is partially explained by the following principles.
Firstly, we have to consider signal specificity. MAP kinases are very active proteins with many targets, and so need to be under close regulation. MEK provides this, since the peculiar phosphorylation requirements of MAP kinases (see below) mean that MEK is the only known activator of MAP kinases. Assembling the three-kinase signalling module on a scaffold, as happens in some MAP kinase pathway situations, can also help maintain specificity.
Secondly, a sequence of kinases gives an opportunity for signal amplification.
Thirdly, signal duration can radically change the signalling outcome (Section 3.8). Indeed, RTKs and Ras are generally inactivated fairly quickly by tyrosine phosphatases and GAPs, respectively, whereas the MAP kinase cascade can remain active for more extended periods of time (Section 3.7).
Finally, the signal needs to be translocated from one part of the cell (the plasma membrane) to another (the nucleus). The MAP kinase pathway does not employ any second messenger molecules to broadcast the signal, but MAP kinase itself is the nearest protein equivalent, translocating to the nucleus once activated.
Box 3 Investigating the activity of signalling molecules
The activity of signalling molecules can be altered by site-directed mutagenes is which has proved very useful in the engineering of constitutively active or inactive versions of kinases. The inactive versions can act in a dominant negative manner within a cell, blocking the downstream pathway, whereas the constitutively active versions can permanently switch on the downstream pathway. These can be very useful tools for investigating signal transduction pathways. The example we shall examine is the mutagenesis of MEK, which is activated by phosphorylation on two nearby serines, Ser 217 and Ser 221, within the activation loop. The experiment, whose results are illustrated in Figure 38, shows the consequences of introducing MEK constructs that have been mutated at these residues by site-directed mutagenesis into cells. The effects of these mutations are assessed by determining the activity of its immediate downstream effector, ERK, by an in vitro kinase assay.
Replacing serine residues with glutamate residues at positions 217 and 221 creates a constitutively active mutant of MEK, which continuously activates ERK.
From your knowledge of amino acid structure, suggest why glutamate instead of serine at positions 217 and 221 creates a constitutively active mutant.
Glutamate is negatively charged, so to some extent it mimics phosphoserine.
Replacing either of these two key serines with alanine creates an interfering mutant of MEK, which blocks the activation of ERK.
Again, suggest why replacing serine with alanine stops MEK functioning normally.
Because the alanine side-chain has no –OH group, it cannot be phosphorylated, so MEK cannot become activated and cannot phosphorylate ERK.
The technique used to introduce the mutant MEK constructs into cells is known as ‘transfection’ (Figure 39a), a technique for which there are several variants. The principle is to induce tissue culture cells to take up a plasmid expression vector containing the gene to be expressed. Cationic lipids are used to promote uptake of plasmid DNA. In this experiment, a plasmid was used in which the mutant MEK gene was downstream of a promoter (SV40 in this instance). This protocol is suitable for experiments that terminate within a day or two (called ‘transient’ transfection). When a cell line that permanently expresses the gene is needed, the plasmid must be maintained and replicated within the cells.
In this case, the expression vector must have the appropriate replication sequences and a selectable marker (such as the resistance gene to an antibiotic like puromycin), so that transfected cells may be selected from the untransfected neighbours; this is known as ‘stable transfection’ (Figure 39b). When short-term effects need to be observed in individual cells, or where cells are difficult to transfect, recombinant DNA (or protein) may be microinjected (Figure 39c).

Figure 40 shows the rat PC12 neural cell line microinjected with MEK mutant DNA. The constitutively active MEK mutant induces differentiation of this cell line (as seen by the outgrowth of neural processes, also known as ‘neurites’), similar to that induced by nerve growth factor (NGF). This evidence supports a role for the ERK MAP kinase pathway in neural outgrowth in this cell type.
The activity of a signalling protein can be altered not only by changing the catalytic activity of its kinase domain, but also by translocating the signalling protein to particular subcellular localizations, which can be critical for their activation and/or ability to access their targets. This can be investigated experimentally by ‘forcing’ the protein of interest into a particular subcellular localization. The principle has been successfully exploited, for example, to investigate the activation of Raf. The membrane localization signal of K-Ras (the amino acid sequence Cys–Val–Ile–Met), which is post-translationally prenylated by addition of a farnesyl residue, was added to the carbon terminus of Raf. This modification constitutively localized Raf to the membrane, resulting in its constitutive activation. The activated Raf could be further activated by EGF independently of Ras, which suggested that Ras acts as a temporary membrane anchor for Raf (as shown in Figure 36), and that Raf is also activated by other membrane-associated signals when in the membrane environment.
Proteins may be targeted to a number of cellular locations by this technique. For example, nuclear localization sequences can be added to signalling proteins (such as MAP kinases) to investigate their role in gene transcription.
What signal sequence do you think would target a protein to the nucleus?
A nuclear localization signal (NLS).
Activation of MAP kinases involves specific phosphorylation of residues within the activation loop of the catalytic domain of the protein, For example, Raf activates MEK by phosphorylating two specific serines within the activation loop (Box 3). The activation requirements of MAP kinase are even more specific. It requires activation by phosphorylation of a threonine residue and a tyrosine residue. These two are separated from the threonine receptor by a single amino acid within the activation loop. In ERK this motif is Thr–Glu–Tyr, in which the threonine and tyrosine residues can be phosphorylated only by MEK, because MEK is a member of the very unusual dual-specificity kinases, which are able to phosphorylate serine–threonine and tyrosine residues.
Activated MAP kinase family members are activators of immediate ‘early genes’, so called because they are activated within minutes of cell stimulation. Many of these genes encode transcription factors (Section 3.8), which then switch on other sets of genes, thereby initiating cellular programs of differentiation or proliferation. There are also cytosolic targets of MAP kinases, which include regulators of protein synthesis. As an example, MAP kinase phosphorylates another kinase called Mnk1, which, in turn, phosphorylates and activates the translation initiation factor 4E (eIF-4E.
There are several MAP kinase pathways. The pathways employing ERK represent the classic pathway, but there are also two other major mammalian MAP kinase pathways implicated in cellular responses to stress (not discussed here). Moreover, the MAP kinase pathway is conserved across the animal kingdom and even in yeast and plants, as shown in Table 3. In particular, the model organisms C. elegans and Drosophila have provided extremely useful experimental systems for investigating MAP kinase pathways, and for showing that it operates downstream of growth factor receptors and Ras.
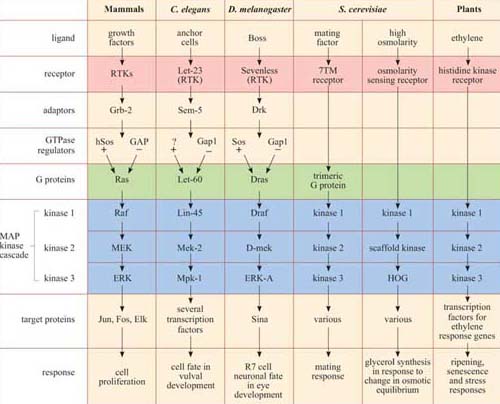
*There is remarkable conservation of MAP kinase signalling pathways, from mammals to plants. In addition to the MAP kinase cascade (blue), there are other striking homologies in several of the pathways, especially between growth factor pathways in mammals, vulval induction in nematodes and eye development in fruit-flies, where the receptors (pink), adaptors, GTPase regulators and G protein homologues (green) are also related. Not all the names of all the signalling proteins here are explained, and they need not be remembered; they are included merely to illustrate the conservation of the pathways. The GEF for the Ras homologue in C. elegans remains unidentified, and is denoted as ‘?’.
3.6.2 The JAK–STAT pathway
Another important protein kinase pathway is the JAK–STAT pathway. Cytokines (Section 2.2), are frequently used for signalling between cells of the immune system. Cytokine-induced signal transduction cascades are often direct pathways to the nucleus for switching on sets of genes. Janus kinases (JAKs, named after the two-faced Roman god) are a particular group of tyrosine kinases that associate with cytokine receptors. When cytokines such as α-interferon bind to their receptor, JAKs (denoted as JAK1, Tyk2, etc.) associated with different receptor units cross-phosphorylate each other on tyrosine residues (Figure 41). The receptors are then phosphorylated by JAKs, creating phosphotyrosine-docking sites for SH2-containing proteins, in this case a specific group of proteins called STATs (signal transducers and activators of transcription).
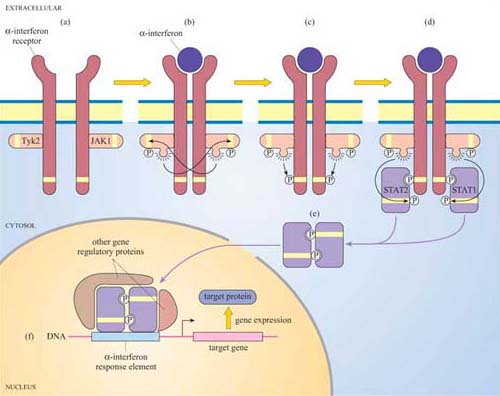
Box 4 Detecting protein–protein interactions using the MAPPIT system
The mammalian protein–protein interaction trap (MAPPIT) system uses the principle, instead of the genes of interest being fused to transcriptional activator domains, they are fused to components of the JAK–STAT signalling pathway (Figure 42). Using recombinant DNA technology, the bait protein domain is fused to a mutant erythropoietin (Epo) receptor, which has JAK binding sites but lacks STAT binding sites. The prey protein is fused to a protein fragment that has several potential STAT binding sites. If there is no interaction between bait and prey, JAKs cross-phosphorylate each other on stimulation with Epo, but STAT is not recruited as there are no available docking sites on the receptor. If there is an interaction between bait and prey, the protein fragment becomes tyrosine-phosphorylated by JAKs on receptor activation, enabling STAT to bind to it. STAT is then in the vicinity of JAK, which can access and phosphorylate the STAT. Then STAT can dimerize, translocate to the nucleus and induce transcription of a reporter gene which can be easily quantified.
In turn, the STATs are phosphorylated by the JAKs, which causes the STATs to dissociate from the receptor and instead bind to each other by means of their SH2 domains. STAT dimers translocate directly to the nucleus, where they bind to other gene regulatory proteins and to response elements in target genes.
Can you recall any other signalling pathway that has also has a direct route to the nucleus?
The nuclear hormone receptor signalling pathway, where the intracellular receptor binds the ligand (for example, a steroid) and translocates to the nucleus, where it binds to hormone response elements (Figure 27).
The JAK–STAT pathway has recently become the basis for the development of techniques for the study of protein–protein interactions (Section 1.5).
Now that we have described the major protein kinases, we can look at them all together, and see what features they have in common. Figure 43 is a dendrogram showing how the various subgroups of protein kinases in humans are related to each other both functionally and structurally. Despite their different subcellular localizations, activation mechanisms and substrates, protein kinases have remarkable similarity of structure within their kinase domain.
3.7 Protein phosphatases
Together with inositolphospholipid phosphatases, protein phosphatases are key regulators of signal transduction pathways. Like protein kinases, protein phosphatases are either tyrosine phosphatases (the majority of protein phosphatases, some of which are shown in Figure 44) or serine–threonine phosphatases (including the phosphoprotein phosphatase family, designated PP1–6), which will be described in Section 4, or, rarely, dual-specificity phosphatases.
Phosphatases are required to inactivate signalling proteins that have been activated by phosphorylation. Many tyrosine phosphatases such as SHP-1 and -2 have SH2 domains, and are recruited to the membrane following ligand-stimulated phosphorylation of receptors. For example, the tyrosine phosphatase SHP-1 binds to phosphotyrosines on activated cytokine receptors such as the erythropoietin (Epo) receptor, and is then phosphorylated by JAK2, which activates it. Active SHP-1 can downregulate (damp down) the JAK/STAT signalling pathway by dephosphorylating specific JAKs and STATs. It is therefore acting as a negative regulator.
Another role for phosphatases occurs when they activate a protein that is held in an inactive state by phosphorylation.
You have already met an example of a protein regulated in this way. What is it?
Src, which in its inactive state has an inhibitory phosphate on Tyr 527 (Section 1.6).
Lck is a Src family tyrosine kinase (Figure 26), which is dephosphorylated and thereby activated by the membrane-bound tyrosine phosphatase CD45. (CD45 plays an essential role in the activation of leukocytes following antigen presentation.) CD45, like many of the PTPs, is a transmembrane protein (Figure 44); such proteins are referred to as receptor tyrosine phosphatases.
One of the most well-studied phosphatases is the dual-specificity phosphatase MKP-1, which inactivates MAP kinase. The MKP-1 gene is one of the immediate early genes expressed following MAP kinase activation, being expressed approximately 20 minutes after cell stimulation. As MKP-1 levels rise, MAP kinase is dephosphorylated and inactivated.
What type of regulation is effected by MKP-1 on MAP kinase?
Feedback inhibition.
Because negative feedback by MKP-1 is a transcription-dependent mechanism, it helps to explain the relatively long duration of MAP kinase activation.
3.8 Activation of transcription factors
We have already come across several examples of signalling pathways leading to activation (or inactivation) of transcription factors, which in turn modulate transcription of sets of genes leading to, for example, programs of differentiation or proliferation. You will also meet several other specific examples in subsequent chapters. For now, we shall examine one particular scenario, namely the activation of immediate early genes by MAP kinases, which illustrates some of the principles and details involved.
One of the most important immediate early genes activated by the ERK family of MAP kinases is fos. The fos gene product is itself an important transcription factor, which helps to activate transcription of genes containing binding sites for the transcription factor complex AP-1 (formed by association of Fos and Jun; Figure 45) in their promoters. ERK brings about transcription of fos by phosphorylating the nuclear transcription factor known as ‘ternary complex factor (TCF)’. This, together with the serum response factor (SRF), binds to the serum response element (SRE) in the promoter of the fos gene and increases its transcription rate (Figure 45).
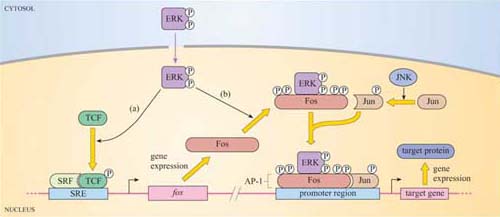
It takes about 30 minutes or more for the proteins encoded by immediate early genes such as fos to be synthesized. Once Fos protein has been produced, MAP kinases participate in its activation, in a stepwise manner, firstly by phosphorylating two sites in the C-terminal region of Fos. This exposes the ERK-binding motif of Fos. ERK then further phosphorylates Fos at two more sites. It is immediately apparent that if MAP kinase activation is only transient, lasting for less than 30 minutes, Fos cannot be activated. Therefore, we can see how transient MAP kinase activation can lead to a different cellular outcome than sustained MAP kinase activation.
This is illustrated by the neural cell line PC12, which fully differentiates them into neurons only when ERK activation is sustained (for example, by addition of nerve growth factor; Figure 40c).
Can you think of an experimental method for the investigation of the relationship between sustained activation of ERK and differentiation of PC12 cells?
By transfection of PC12 cells with constitutively active mutant forms of MEK and analysing neurite outgrowth (Box 3).
3.9 Summary
Heterotrimeric G proteins are tethered to the internal surface of the plasma membrane, and are activated by conformational change within 7TM receptors. There are many different α subunits (and a few βγ subunits), which interact with different receptors and different effectors. The major targets of G proteins include ion channels, adenylyl cyclase (activated by Gαs and inhibited by Gαi) and PLC-β (activated by Gαq).
Phosphatidylinositol (PI) is the precursor of a family of small lipid second messengers. The inositol ring can be further phosphorylated at positions 3, 4, and 5 by lipid kinases. PI 3-kinase specializes in phosphorylating the hydroxyl group at the 3 position, thus generating the active signalling molecules PI(3,4)P2 and PI(3,4,5)P3. The phosphorylated 3 position is recognized as a docking site by PH domain-containing proteins, thus providing a mechanism for signalling proteins to be recruited to the membrane.
Phospholipase C enzymes (especially PLC-β, activated by G proteins, and RTK-activated PLC-γ) cleave PI(4,5)P2 to generate diacylglycerol (DAG) and inositol 1,4,5-triphosphate (IP3). DAG remains embedded in the membrane, where it activates protein kinase C (PKC). IP3 diffuses through the cytosol, and opens IP3-gated calcium channels, releasing stored calcium into the cytosol.
The Ca2+ ion is an important second messenger, which enters the cytosol from the extracellular space through specific channels on the plasma membrane, or is rapidly released from stores into the cytoplasm. Calcium channels include IP3-gated calcium channels, voltage-dependent calcium channels, or ryanodine receptors in skeletal muscle cells. It activates numerous Ca2+ -dependent proteins, including PKC, but many of its effects are mediated via calmodulin, which has four allosteric Ca2+ binding sites. Ca2+ /calmodulin then binds to and regulates target proteins, especially Ca2+ /calmodulin-dependent protein kinases (CaM kinases).
Cyclic AMP (cAMP) is another important second messenger, synthesized by adenylyl cyclase (which is activated or inhibited by different G protein subtypes). It can open cAMP-gated ion channels, but it mediates many of its effects through cAMP-dependent protein kinase A (PKA), whose roles include regulating glycogen metabolism, and phosphorylation of a transcription factor that binds to the cAMP response element (CRE).
Cyclic GMP is synthesized by guanylyl cyclase. Its targets include cGMP-gated ion channels and a cGMP-dependent kinase (PKG).
Ras is the archetypal monomeric, or small, G protein. Ras classically operates downstream of growth factor receptors: Grb-2, an SH2/SH3-containing adaptor protein, binds to phosphotyrosines on the activated RTK, and recruits Sos to the membrane environment; Sos promotes GTP binding by Ras. Activated Ras has more than one target, including PI 3-kinase, but its most important downstream pathway is the MAP kinase pathway. Activated Ras recruits Raf to the membrane, where it is activated and then phosphorylates MEK, which then phosphorylates ERK, a MAP kinase. These have multiple cytoplasmic and transcription factor targets involved in cell growth and division or differentiation.
Protein kinase families involved at various points in signalling pathways include receptor tyrosine kinases (for example, the EGF receptor), non-receptor tyrosine kinases (such as Src and JAK), serine–threonine kinases such as PKC, PKA, MAP kinases and the TGF receptor, and rare dual-specificity kinases such as MEK.
Protein phosphatases dephosphorylate proteins, and are grouped according to their targets, as are protein kinases. They include protein tyrosine phosphatases, serine–threonine phosphatases, and a few dual-specificity phosphatases.
The duration of ERK activity determines activation of different transcription factors (SRF/TCF for immediate early genes; AP-1 for other target genes).
The major signalling molecules we have discussed are brought together into five basic pathways in Figure 46.
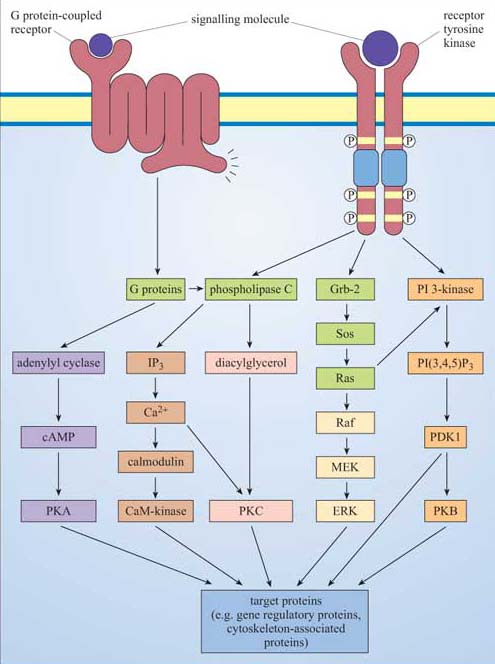
4 Glucose metabolism: an example of integration of signalling pathways
4.1 Glucose metabolism
We are now in a position to draw together the major concepts and components of signalling, and show how they operate in one well-understood system, namely the regulation of the storage or release of glucose in the human body. From this, you will be able to recognize archetypal pathways represented in specific examples, you will be able to appreciate how the same basic pathways can be stimulated by different hormones in different tissues, and you will see how opposing hormones activate separate pathways that affect the same targets but in opposite ways.
Following a meal, insulin is released into the bloodstream by pancreatic β cells. The overall systemic effects of insulin are to increase uptake of blood glucose into cells, and to promote its storage as glycogen in muscle and liver cells. (Note that glycogen is a polysaccharide consisting of repeated units of glucose used for shortterm energy storage by animal cells.) A rise in the concentration of blood glucose, such as that following the consumption of food, stimulates insulin production, which signals through the insulin RTK. The insulin RTK phosphorylates various substrate proteins, which link to several key signalling pathways such as the Ras–MAP kinase pathway. There are, however, two major pathways that control glycogen synthesis and breakdown in animal cells (Figure 47).
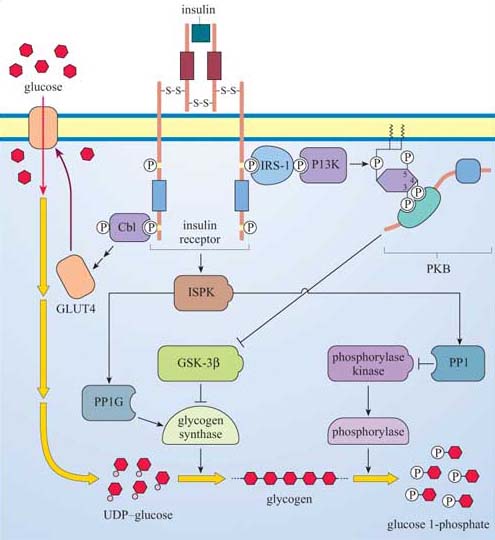
Glygogen synthase kinase-3β (GSK-3β) inhibits glycogen synthase (GS), which is responsible for synthesizing glycogen from uridine diphosphate (UDP)–glucose.
Phosphorylase kinase, a serine–threonine kinase of the CaM kinase family, which, in turn, activates the enzyme phosphorylase, responsible for cleaving glucose 1-phosphate units from the glycogen chain.
Both regulatory mechanisms are influenced by signalling cascades initiated by the interaction between phosphotyrosine residues on the insulin receptor and two signalling molecules:
The first is Cbl, which was introduced in Section 2.4 as a link between RTKs and ubiquitin in receptor sequestration. In this case, it seems to be the start of a pathway that ends up with translocation of GLUT4 glucose-transporter molecules to the plasma membrane, thus promoting the uptake of glucose into the cell.
The second is IRS-1, insulin receptor substrate-1, which is a large protein that binds several SH2-containing proteins, including PI 3-kinase. As described previously (Section 3.3), PI 3-kinase creates phosphorylated inositol phospholipid docking sites for PH-containing proteins, including PKB. GSK-3β is inhibited by PKB, so that its inhibitory action on glycogen synthase activity is negated.
At the same time, an insulin-stimulated protein kinase (ISPK, activated by insulin by an unknown mechanism) acts on the serine–threonine phosphatase PP1G to further enhance glycogen synthase activity. ISPK also activates another serine–threonine phosphatase, PP1, which negatively regulates the activity of phosphorylase kinase, and, ultimately, phosphorylase.
So insulin promotes uptake of glucose into tissues by mobilizing glucose transporters, and in liver and skeletal muscle, activates glycogen synthesis by modulating the activity of GSK-3β and GS, and inhibits glycogen breakdown via PP1. In other words, insulin induces gluconeogenesis in liver and skeletal muscle.
Two hormones, adrenalin (secreted from the adrenal medulla in anticipation of muscle activity) and glucagon (released from pancreatic α cells when blood sugar is low) have the opposite effect to insulin; that is, they increase the rate at which glycogen is converted to glucose (glycogenolysis; Figure 20). In skeletal muscle, glucose enters the glycolytic pathway to produce ATP, the fuel for muscle contraction. In the liver (which is more responsive to glucagon than to adrenalin), glucose is released into the bloodstream for use by muscle cells. Adrenalin and glucagon act through GPCRs, so their signalling pathways start off quite differently from that of insulin, a RTK. However, they end up (among other things) regulating the activity of the same enzymes involved in glycogen synthesis that insulin itself modulates.
Adrenalin has many effects, but in skeletal muscle it acts through β-adrenergic receptors, which are coupled to Gαs and stimulate adenylyl cyclase activity. This results in cAMP elevation and consequently activation of PKA (Figure 48). PKA activates phosphorylase kinase (see (a) in Figure 48), which in turn activates the enzyme phosphorylase. (Phosphorylase kinase activation is also promoted by ACh release from neuron terminals; Figure 48). PKA also phosphorylates three key proteins to promote glycogen breakdown and inhibit its synthesis as shown by letters (b)–(d) in Figure 48:
(b) The serine–threonine phosphatase PP1, which is inhibited as a result (PP1's action is to dephosphorylate, and therefore inhibit, phosphorylase kinase and phosphorylase). Note that PKA phosphorylates (and inactivates) PP1 on different residues than ISPK (which activates PP1).
(c) PP1 inhibitor, which on phosphorylation is activated.
(d) Glycogen synthase, which on phosphorylation is inactivated.
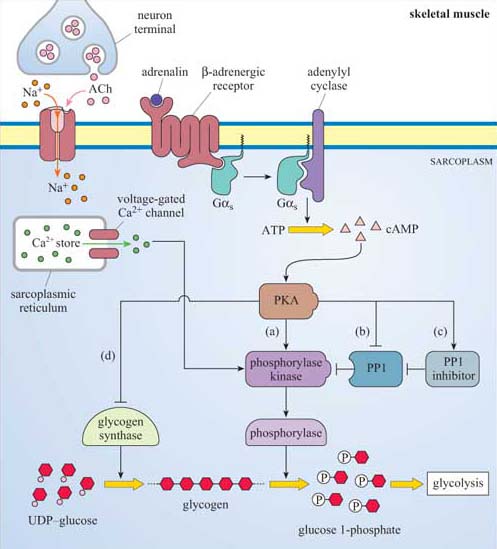
This system provides a good illustration of how a key signalling enzyme (PKA) with multiple substrates can regulate different targets within the same metabolic pathway, all combining to promote one outcome, in this case glycogen breakdown. In at least two cases (glycogen synthase and PP1), it is the same enzymes whose activity is ultimately regulated (either negatively or positively) by insulin (via PKB) and adrenalin (via PKA), acting antagonistically.
In the liver, however, adrenalin acts mainly but not exclusively through α-adrenergic receptors, which are coupled to Gαq, and activate the PLC/IP3 pathway, which opens IP3-gated Ca2+ channels (Figure 49), helping to activate phophorylase kinase and promote glycogen breakdown, leading to glucose release into the blood.
Glucagon is a hormone that activates glycogen breakdown, particularly in the liver, resulting in a release of glucose into the blood. In the liver, the control of glycogen breakdown is fundamentally the same as in skeletal muscle, but with particular differences. Whereas skeletal muscle needs to be extremely responsive to adrenalin (for the classic fight or flight response), the function of the liver is to maintain blood sugar levels within a constant, physiological range. Liver cells therefore have many glucagon receptors (Figure 49), which are GPCRs coupled to Gαs, activating the same basic pathway that adrenalin does in skeletal muscle, via cAMP/PKA. The result is phosphorylation and activation of phosphorylase kinase, thereby promoting glycogen breakdown.
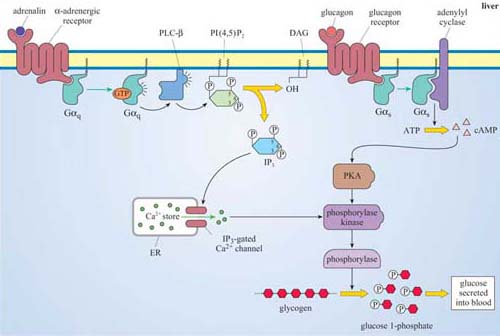
4.2 Summary
Glycogen metabolism is controlled by two enzymes, glycogen synthase (mediating glycogen synthesis) and phosphorylase (mediating glycogen breakdown).
Three pathways converge in the regulation of glycogen synthase: cAMP/PKA and GSK-3β are negative regulators, whereas ISPK/PP1G positively regulate the activity of glycogen synthase.
Insulin and adrenalin have opposite effects on glycogen synthesis: insulin promotes glycogen synthesis by activating ISPK/PP1G and by inhibiting GSK-3β by the PI3K/PKB pathway, whereas adrenalin inhibits glycogen synthase by the cAMP/PKA pathway.
Three pathways converge in the activation of phosphorylase by phosphorylase kinase: Ca2+ and PKA activate phosphorylase kinase, whereas PP1 is a negative regulator.
Acetylcholine, adrenalin and glucagon promote glycogen breakdown, whereas insulin inhibits it.
Acetylcholine in skeletal muscle and adrenalin in liver activate phosphorylase kinase by a common mechanism, an increase in cytosolic Ca2+, although the effect of ACh is by voltage-dependent channels and that of adrenalin by IP3-gated Ca2+ channels.
Adrenalin in muscle and glucagon in liver activate phosphorylase kinase by a common mechanism, namely an increase in cytosolic cAMP and subsequent activation of PKA. In addition, PKA further activates phosphorylase kinase in skeletal muscle by inhibition of PP1 either directly or indirectly.
Insulin has an opposite effect to adrenalin on glycogen breakdown, namely the inhibition of glycogen breakdown by activation of the phosphorylase kinase inhibitor, PP1.
Conclusion
This free course provided an introduction to studying Science. It took you through a series of exercises designed to develop your approach to study and learning at a distance and helped to improve your confidence as an independent learner.
References
Acknowledgements
Grateful acknowledgement is made to the following sources for permission to reproduce material in this course:
This content is made available under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 LicenceSee Terms and Conditions
Course image: rosipaw in Flickr made available under Creative Commons Attribution-NonCommercial-ShareAlike 2.0 Licence.
Figure: 1 Copyright © Michael Snyder
Figure 2, 6, 11, 12, 23, 25, 27, 30a, b, c, 32, 41, 44, 46 Alberts, B. et al. (2002) Molecular Biology of the Cell, 4th edn, Garland Science, reproduced with permission of Routledge/Taylor and Francis Books, Inc
Figure 9 Jhun, B.H. (1995) ‘The MATK tyrosine kinase interacts in a specific and SH2-dependant manner with c-Kit’, Journal of Biological Chemistry, 270 (16), The American Society for Biochemistry and Molecular Biology
Figure 21b, c, d Courtesy of Nigel Unwin, MRC Laboratory of Molecular Biology, Cambridge
Figure 21e, 13.22 Gomberts, B.D. et al (2002) Signal Transduction, Academic Press
Figures 38, 40 Cowley, S. et al (1994) ‘Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells’, Cell 77, pp. 841-852, copyright © 1994, with permission from Elsevier
Figure 4 Eycherman, S. et al (2001) ‘Design and application of cytokine-receptor-based interaction trap’, Nature Cell Biology, 3(12)
Table 1 Gomperts, B.D. et al (2002) Signal Transduction, Academic Press
All other material contained within this course originated at the Open University
Don't miss out:
If reading this text has inspired you to learn more, you may be interested in joining the millions of people who discover our free learning resources and qualifications by visiting The Open University - www.open.edu/ openlearn/ free-courses