Influenza: A case study
Use 'Print preview' to check the number of pages and printer settings.
Print functionality varies between browsers.
Printable page generated Wednesday, 3 June 2026, 5:18 PM
Influenza: A case study
Introduction
Most people have suffered from influenza (flu) at some time in their lives, so you probably have personal experience or a good idea of the symptoms and progression of the disease. However, influenza is actually one of the world’s most serious diseases. The pandemic of flu that occurred in 1918, immediately following the First World War, is thought to have killed up to 50 million people: many more than died in the war itself. More recently, the 2009 ‘swine flu’ pandemic caused widespread panic across parts of the world, although it resulted in relatively few fatalities.
The following course is a case study of influenza that considers a range of topics such as the nature of the virus itself, its spread, treatment, and diagnosis. There are activities to complete and videos to watch as you work through the text, and a set of self-assessment questions at the end of the course that allow you to judge how well you have understood the course content.
This OpenLearn course is an adapted extract from the Open University course : SK320 Infectious disease and public health.
Learning outcomes
After studying this course, you should be able to:
define and use, or recognise definitions and applications of, each of the glossary terms in the course
describe influenza viruses, their structure, how they are transmitted, how they infect cells and replicate and how they produce their damage in the host
outline the different types of immune defence which are deployed against flu infections, distinguishing those that act against infected cells from those that act against free virus
describe how strains of the virus change over time, and relate this to the flu viruses that occur in birds and other mammals
explain how the epidemic pattern of influenza can be related to the evolution of new strains of virus and to the specificity of the immune response against each strain.
1 Background to the case study
Influenza is a myxovirus belonging to the family of viruses known as Orthomyxoviridae. The virus was originally confined to aquatic birds, but it made the transition to humans 6000–9000 years ago, coinciding with the rise of farming, animal husbandry and urbanisation.
These changes in human behavior and population density provided the ecological niche that enabled influenza, as well as a number of other infectious agents such as the viruses that cause measles and smallpox, to move from animals and adapt to a human host.
Influenza as a disease has been recognised for centuries, even though the viruses which cause it were not correctly identified until the early 1930s, first in the UK and then in the USA. Indeed the name itself is derived from an Italian word meaning ‘influence’, and reflected the widespread belief in medieval times that the disease was caused by an evil climatic influence due to an unfortunate alignment of the stars.
Our current understanding – that infectious diseases are caused by infectious agents – is so ingrained that such mystical causes for an illness now seem absurd. However, even during the Middle Ages, people had a sound idea of infection and realised that some diseases could be passed from one individual to another and others could not. For example, the use of quarantine for a disease such as plague, but not for many other illnesses, shows that people could distinguish infectious diseases from non-infectious diseases even if the causative agent and the method of transmission were obscure.
The idea that influenza is caused by the influence of the stars, though not a satisfactory explanation of how the disease spread, does identify an important feature of flu – that serious epidemics of the disease occur at irregular intervals.
For example, in the twentieth century there were at least five major epidemics of flu that spread around the world (a pandemic), and there were less serious epidemics in most years. In times when people believed in the spontaneous generation of life, the stars would have seemed a reasonable explanation for unpleasant and unexpected epidemics.
1.1 Defining influenza
How would you define ‘influenza’?
You may well have defined influenza as an infection caused by an influenza virus. However, you may have defined it according to its symptoms: an infection that starts in the upper respiratory tract, with coughing and sneezing, spreads to give aching joints and muscles, and produces a fever that makes you feel awful; but usually it has gone in 5–10 days and most people make a full recovery.
The first answer here is the biological definition and, in the Open University course SK320, diseases are defined according to the infectious agent which produces them. This is because different infections can produce the same symptoms, and the same infectious agent can produce quite different symptoms in different people, depending on their age, genetic make-up or the tissue of the body that becomes infected. Here a distinction is made between the infectious disease caused by a particular agent and the disease symptoms.
Unfortunately there is a lot of confusion in common parlance about different diseases. Often, people say that they have ‘a bit of flu’ when they have an infection with some other virus, or a bacterium that produces flu-like symptoms. Such loose terminology is understandable, since most people are firstly concerned with the symptoms of their disease. But to treat and control disease requires accurate identification of the causative agent, so this is the starting point for considering any infectious disease.
Attributing cause to a disease
The difficulties encountered in assigning a particular pathogen to a disease are well-illustrated by influenza.
During the influenza pandemic that occurred in 1890, the microbiologist Pfeiffer isolated a novel bacterium from the lungs of people who had died of flu. The bacterium was named Haemophilus influenzae and since it was the only bacterium that could be regularly cultivated from these individuals at autopsy, it was assumed that H. influenzae was the causative agent of flu.
Again, in the 1918 flu pandemic, the bacterium could be regularly cultivated from people who had died of flu with pneumonia. So it was thought that flu was caused by the bacterium, and H. influenzae came to be called the ‘influenza bacillus’.
The role of H. influenzae was only brought into question in the early 1930s, when Smith, Andrews and Laidlaw showed that it was possible to transfer a flu-like illness from the nasal washings of an infected person to ferrets, using a bacteria-free filtrate. These studies demonstrated that the pathogen was in fact much smaller than any known bacterium and paved the way to the identification of influenza viruses (Figure 1).
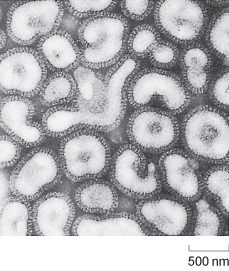
Why do you suppose that H. influenzae was incorrectly identified as the causative agent of flu?
The bacterium fulfils two of Koch’s postulates: it is regularly found in serious flu infections and it can be cultured in pure form on artificial media. Moreover, at that time no-one knew what a virus was, and everyone was thinking in terms of bacterial causes for infectious diseases.
Although the precise role of H. influenzae in the 1890 and 1918 flu pandemics is not clear, it is likely that the bacteria were present and acting in concert with the flu virus to produce the pneumonia experienced. Such synergy between virus and bacteria was demonstrated by Shope in 1931. He infected pigs with a bacterial-free filtrate (containing swine influenza virus) with or without the bacteria, and showed that the disease produced by the bacteria and filtrate together was more severe than that produced by either one alone (Van Epps, 2006).
In its role of co-pathogen, H. influenzae is only one of a number of bacteria that can exacerbate the viral infection. This highlights a very important point. In the tidy world of a microbiology or immunology laboratory, scientists typically examine the effect of one infectious agent in producing disease. In the real world, people often become infected with more than one pathogen. Indeed, infection with one agent often lays a person open to infection with another, as immune defences become overwhelmed. For this reason, a particular disease as seen by physicians may be due to a combination of pathogens.
1.2 Influenza infection in humans
Influenza is an acute viral disease that affects the respiratory tract in humans. The virus is spread readily in aerosol droplets produced by coughing and sneezing, which are symptoms of the illness. Other symptoms include fatigue, muscle and joint pains and fever.
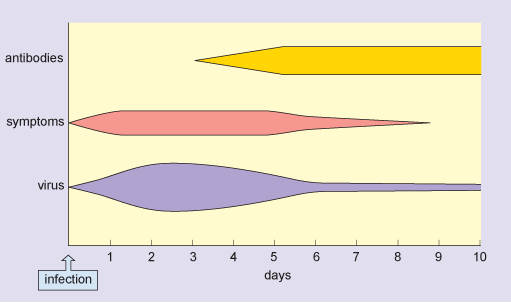
Following infection, the influenza virus replicates in the cells lining the host’s upper and lower respiratory tract. Virus production peaks 1–2 days later, and virus particles are shed in secretions over the following 3–4 days. During this period, the patient is infectious and the symptoms are typically at their most severe.
After one week, virus is no longer produced, although it is possible to detect viral antigens for up to 2 weeks. Immune responses are initiated immediately after the virus starts to replicate, and antibodies against the virus start to appear in the blood at 3–4 days post infection. These continue to increase over the following days and persist in the blood for many months.
In a typical flu infection, the virus is completely eliminated from the host’s system within 2 weeks. This is sterile immunity: the virus cannot be obtained from the patient after recovery from the disease. Figure 2 shows the typical time course of an acute flu infection.
For infants, older people, and those with other underlying diseases (e.g. of the heart or respiratory system) an infection with flu may prove fatal. However, the severity of a flu epidemic and the case fatality rate depend on the strain of flu involved and the level of immunity in the host population.
During a severe epidemic, there are typically thousands more deaths than would normally be expected for that time of year, and these can be attributed to the disease. Although older people are usually most at risk from fatal disease, this is not always so. In the 1918 flu pandemic there was a surprisingly high death rate in people aged 20–40 (Figure 3), and this was also the case for the 2009 ‘swine flu’ pandemic.
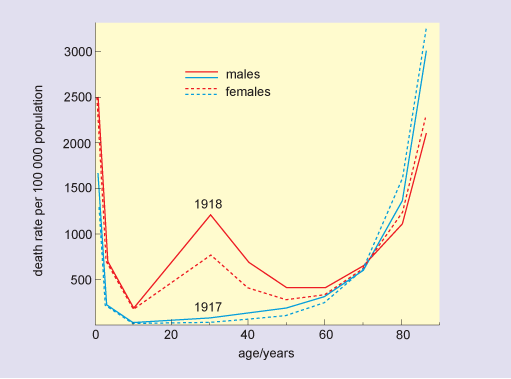
Older people are often most severely affected during infectious disease outbreaks because they may have a less effective immune response than younger people, or a reduced capacity to repair and regenerate tissue damaged by the infection. However, there are circumstances where older people may be more resistant to infection than younger people because they may have already encountered the disease (in their youth) and could retain some immunity and so be less susceptible than younger people who have not encountered the disease before.
1.3 Influenza infection in other species
Influenza viruses infect a wide range of species, including pigs, horses, ducks, chickens and seals. In most of these other species the virus produces an acute infection.
For example, in most of the mammals the symptoms are very similar to those in humans: an acute infection of the respiratory tract, which is controlled by the immune response although fatal infections occur in some species. However, in wild ducks and other aquatic birds the virus primarily infects the gut and the birds do not appear to have any physical symptoms.
Despite this, ducks may remain infected for 2–4 weeks and during this time they shed virus in their faeces. Potentially this is a very important reservoir of infection; although flu viruses do not often cross the species barrier, the pool of viruses present in other species is an important genetic reservoir for the generation of new flu viruses that do infect humans.
This reservoir becomes particularly important in certain farming communities or in crowded conditions where animals (especially pigs and ducks) are continuously in close proximity with humans (Branswell, 2010). Although such conditions occur in many agricultural communities throughout the world, they are typically observed in South-East and East Asia thereby contributing to these geographical areas often being the source of radically new ‘hybrid’ strains of influenza that incorporate genes from different species-specific strains. (The genetics of influenza are discussed in Section 2.3.)
When strategies for controlling a disease are considered, awareness of the possible presence of an animal reservoir of infection is very important. For example, an immunisation programme against flu would substantially reduce the incidence of the current strain in humans but, because there is always a reservoir of these viruses in other animals, and these viruses are constantly mutating, another strain would inevitably emerge and be unaffected by immunisation. It is useful to distinguish diseases such as rabies, which primarily affect other vertebrates and occasionally infect humans (zoonoses), from diseases such as flu where different strains of the virus can affect several species including humans.
Identify a fundamental difference between the way that zoonoses (e.g. rabies) are transmitted, and the way in which flu is transmitted.
Flu can be transmitted from one human being to another, whereas most zoonoses, including rabies, are not transmitted between people.
2 Influenza viruses
Viruses have very diverse genomes. Whereas the genomes of bacteria, plants and animals are of double-stranded DNA, the genomes of viruses can be constituted from either DNA or RNA and may be double- or single-stranded molecules.
Usually, DNA is a double-stranded molecule with paired, complementary strands (dsDNA) and RNA is a single stranded molecule (ssRNA). However, some viruses have single-stranded DNA genomes (ssDNA) and some have double-stranded RNA genomes (dsRNA). The type of nucleic acid found in the genome depends on the group of viruses involved.
RNA encodes protein in all living things, and the sequence of bases in the RNA determines the sequence of amino acids in the protein. A strand of RNA which has the potential to encode protein is said to be ‘positive sense’ (+). If a strand of RNA is complementary to this, then it is ‘negative sense’ (–). Negative-sense RNA must first be copied to a complementary positive-sense strand of RNA before it can be translated into protein.
The description of the influenza genome as negative-sense ssRNA means that its RNA cannot be translated without copying first. This copying is performed by influenza’s viral RNA polymerase, a small amount of which is packaged with the virus, ready to begin copying the viral genome once it enters a host cell. Viral RNA polymerase consists of three subunits: PB1, PB2, and PA, encoded separately by the first three viral RNA strands.
Understanding the way in which different viruses replicate is important, since it allows the identification of particular points in their life-cycle that may be susceptible to treatment with antiviral drugs.
Classification
Viruses are classified into different families, groups and subgroups in much the same way as are species of animals or plants.
As you have already read, the influenza viruses are (–)ssRNA organisms (Baltimore group V) and belong to a family called the Orthomyxoviruses (see Box 1). They fall into three groups: influenza A, B and C.
Type A viruses are able to infect a wide variety of endothermic (warm-blooded) animals, including mammals and birds, and analysis of their viral genome indicates that all strains of influenza A originated from aquatic birds.
By contrast, types B and C are mostly confined to humans. At any one time, a number of different strains of virus may be circulating in the human population.
Box 1 Families, groups and strains of virus
Viruses were originally classified into different groups according to similarities in their structure, mode of replication and disease symptoms. For example, the Orthomyxoviruses include viruses that cause different types of influenza, while Paramyxoviruses include the viruses that cause measles and mumps.
Such large groupings are often called a family of viruses. The families can be subdivided into smaller groups, such as influenza A, B and C. Even within a single such group of viruses there can be an enormous level of genetic diversity, and this is the basis of the different strains. As an example, two HIV particles from the same individual may be 4% different in their genome; compare this with the 1% difference between the genomes of humans and chimpanzees, which are different species.
2.1 Structure of influenza
The structure of influenza A is shown schematically in Figure 4. The viral genomic RNA, which consists of eight separate strands (see Section 2.3), is enclosed by its associated nucleoproteins to make a ribonucleoprotein complex (RNP), and this is contained in the central core of the virus (the capsid).
The nucleoproteins are required for viral replication and packing of the genome into the new capsid, which is formed by M1-protein (or matrix protein). The M1-protein is the most abundant component of the virus, constituting about 40% of the viral mass; it is essential for the structural integrity of the virus and to control assembly of the virus.
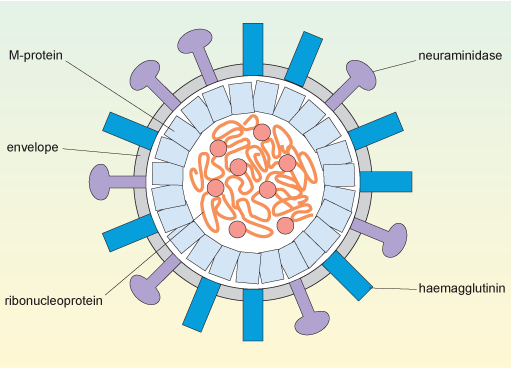
Orthomyxoviruses have a capsid surrounded by a phospholipid bilayer derived from the plasma membrane of the cell that produced the virus. This layer is shown in Figure 4 as the virus’s envelope.
Two proteins, haemagglutinin and neuraminidase, are found on the viral envelope. These proteins are encoded by the viral HA and NA genes (Section 2.3), respectively and are inserted into the plasma membrane of the infected cell before the newly-produced viruses bud off from the cell surface.
The haemagglutinin can bind to glycophorin, a type of polysaccharide that contains sialic acid residues, and which is present on the surface of a variety of host cells. The virus uses the haemagglutinin to attach to the host cells that it will infect. Antibodies and drugs against haemagglutinin are therefore particularly important in limiting the spread of the virus, since they prevent it from attaching to new host cells.
Neuraminidase is an enzyme that cleaves sialic acid residues from polysaccharides. It has a role in clearing a path to the surface of the target cell before infection, namely, digesting the components of mucus surrounding epithelial cells in the respiratory system. Similarly, neuraminidase also promotes release of the budding virus from the cell surface after infection.
The structures of influenza B and influenza C are broadly similar to that of Type A, although in influenza C the functions of the haemagglutinin and the neuraminidase are combined in a single molecule, haemagglutinin esterase. This molecule binds and cleaves a less common type of sialic acid. Influenza C does not normally cause clinical disease or epidemics, so the following discussion is confined to influenza A and B.
2.2 Designation of strains of influenza
A considerable number of genetically different strains of influenza A have been identified, and these are classified according to where they were first isolated and according to the type of haemagglutinin and neuraminidase they express. For example ‘A/Shandong/9/93(H3N2)’ is an influenza A virus isolated in the Shandong province of China in 1993 – the ninth isolate in that year – and it has haemagglutinin type 3 and neuraminidase type 2.
At the start of the twenty-first century, the major circulating influenza A strains are H1N1 (‘swine flu’) and H3N2. At least 16 major variants of haemagglutinin and 9 variants of neuraminidase have been recognised, but to date most of these have only been found in birds.
The designation for influenza B is similar, but omits the information on the surface molecules, for example: ‘B/Panama/45/90’.
As you will see later, accurate identification of different strains of flu is crucial if we are to control epidemics by vaccination programmes.
2.3 Genomic diversity of influenza
The genome of flu viruses consists of around 14 000 nucleotides of negative-sense single-stranded RNA. Compare this number to the approximately 3 billion nucleotides found in the human genome or the 150 billion nucleotides of the genome of the marbled lungfish (the largest genome known in vertebrates).
The genome of influenza viruses is segmented, into eight distinct fragments of RNA containing 11 genes and encoding approximately 14 proteins (see Table 1 below). This structure has significance for the spread of the virus and the severity of disease symptoms.
Cases of influenza generally arise in two main ways: by provoking seasonal annual outbreaks or epidemics and, less commonly, through global pandemics. As you will see shortly, both of these phenomena occur as consequences of the fact that the virus uses RNA as its genetic template and that this RNA genome is segmented into discrete strands.
| Gene name | RNA strand (segment number) | Function(s) of protein encoded by this gene |
|---|---|---|
| PB2 (polymerase basic 2) | 1 | A subunit of viral RNA polymerase involved in cleaving the cap structure of host cell mRNA and generating primers that are subverted for use in the synthesis of viral RNA. |
| PB1 (polymerase basic 1) | 2 | Core subunit of viral RNA polymerase. Required for polymerase assembly. |
| PB1-F2 | 2 | Binds to components of the host mitochondria, sensitising the cell to apoptosis and contributing to pathogenicity. |
| PA (polymerase acidic) | 3 | A subunit of viral RNA polymerase which also has protease activity of unknown function. |
| HA (haemagglutinin) | 4 | Antigenic glycoprotein used for binding to (infecting) the host cell. |
| NP (nucleoprotein) | 5 | RNase resistant protein. Binds viral genomic RNA to form stable ribonucleoproteins and targets these for export from the host nucleus into the cytosol. Also involved in viral genome packaging and viral assembly. |
| NA (neuraminidase) | 6 | Cleaves sialic acid. Important for releasing viral particles from host cell. |
| M1 (matrix 1) | 7 | Binds viral genomic RNA and forms a coat inside the viral envelope in virions. Inside the host cell, it starts forming a layer under patches of the membrane rich in viral HA, NA, and M2 and so facilitates viral assembly and budding from the host cell. |
| M2 (matrix 2) | 7 | Transmembrane ion channel protein. Allows protons into the virus capsid, acidifying the interior, destabilising binding of M1 to the viral genomic RNA which leads to uncoating of the viral particle inside the host cell. |
| NS1 (non-structural 1) | 8 | Inhibits nuclear export of the host’s own mRNA, thereby giving preference to viral genomic RNA. Blocks the expression of some host inflammatory mediators (interferons) and interferes with T cell activation*. |
| NS2/NEP (non-structural 2/ nuclear export protein) | 8 | Mediates the export of viral genomic RNA from the host nucleus to the cytoplasm. |
Footnotes
* Interferons and T cells are involved in the immune response to pathogens.The influenza virus is a successful pathogen because it is constantly changing. How might having a segmented genome promote the evolution of new strains of influenza virus?
If a cell is infected with more than one strain of virus at the same time, then a new strain can be generated simply by mixing RNA strands from different viruses.
2.3.1 Creation of new viral strains
Part of the success of influenza as a pathogen is because segmented genome improves the virus’s potential to evolve into new strains through the combination of different RNA stands. This mixing of the genetic material from different viral strains to produce a new strain is termed genetic reassortment.
For instance, the virus that caused the 2009 H1N1 ‘swine flu’ pandemic comprises a quadruple reassortment of RNA strands from two swine virus, one avian virus, and one human influenza virus:
- the surface HA and NA proteins derive from two different swine influenzas (H1 from a North American swine influenza and N1 from a European swine influenza)
- the three components of the RNA polymerase derive from avian and human influenzas (PA and PB2 from the avian source, PB1 from the human 1993 H3N2 strain)
- the remaining internal proteins derive from the two swine influenzas (MacKenzie, 2009).
This does not necessarily mean that all four viruses infected the same animal at once. The new strain was likely the result of a reassortment of two swine influenza viruses, one from North America and one from Europe. The North American virus may itself have been the product of previous reassortments, containing a human PB1 gene since 1993 and an avian PA and PB2 genes since 2001. The presence of avian influenza RNA polymerase genes in this virus was especially worrying, since the avian polymerase is thought to be more efficient than human or swine versions, allowing the virus to replicate faster and thus making it more virulent. Similar avian RNA polymerase genes are what make H5N1 bird flu extremely virulent in mammals and what made the 1918 human pandemic virus so lethal in people.
This mixing of genes from two or more viruses (whether from the same host species or from different species) can cause major changes in the antigenic surface proteins of a virus, such that it is no longer recognised by the host’s immune system. This antigenic shift is described in more detail in Section 3 (specifically, Box 2).
In contrast to the major genetic changes caused by reassortment, influenza viruses also undergo constant, gradual, genetic changes due to errors made by their RNA polymerases.
2.4 Infection and replication
Influenza RNA polymerase lacks the ability to recognise and repair any errors that occur during genome duplication, resulting in mistakes in copying its viral RNA about once in every 10 000 nucleotides. Because the influenza genome only contains approximately 14 000 nucleotides, this means that, on average, each new virus produced differs by 1 or 2 nucleotides from its ‘parent’.
The slow accumulation of random genetic changes, especially in the antigenic surface proteins, explains why antibodies that were effective against the virus one year may be less effective against it in subsequent years. This gradual change in the nature of viral antigens is known as antigenic drift.
The replication cycle of influenza is illustrated in Figure 5.
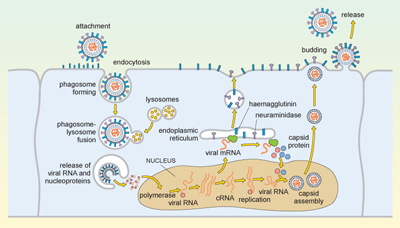
Influenza is spread in aerosol droplets that contain virus particles (or by desiccated viral nuclei droplets), and infection may occur if these come into contact with the respiratory tract. Viral neuraminidase cleaves polysaccharides in the protective mucus coating the tract, which allows the virus to reach the surface of the respiratory epithelium.
The haemagglutinin now attaches to glycophorins (sialic-acid-containing glycoproteins) on the surface of the host cell, and the virus is taken up by endocytosis into a phagosome. Acidic lysosomes fuse with the phagosome to form a phagolysosome and the pH inside the phagolysosome falls. This promotes fusion of the viral envelope with the membrane of the phagolysosome, triggering uncoating of the viral capsid and release of viral RNA and nucleoproteins into the cytosol.
The viral genomic RNA then migrates to the nucleus where replication of the viral genome and transcription of viral mRNA occur. These processes require both host and viral enzymes. The viral negative-stranded RNA is replicated by the viral RNA-dependent RNA polymerase, into a positive-sense complementary RNA (cRNA), and these positive and negative RNA strands associate to form double-stranded RNA (dsRNA). The cRNA strand is subsequently replicated again to produce new viral genomic negative-stranded RNA. Some of the cRNA is also processed into mRNA for translation of viral proteins. The infection cycle is rapid and viral molecules can be detected inside the host cell within an hour of the initial infection.
The envelope glycoproteins (haemagglutinin and neuraminidase) are translated in the endoplasmic reticulum, processed and transported to the cell’s plasma membrane. The viral capsid is assembled within the nucleus of the infected cell. The capsid moves to the plasma membrane, where it buds off, taking a segment of membrane containing the haemagglutinin and neuraminidase, and this forms the new viral envelope.
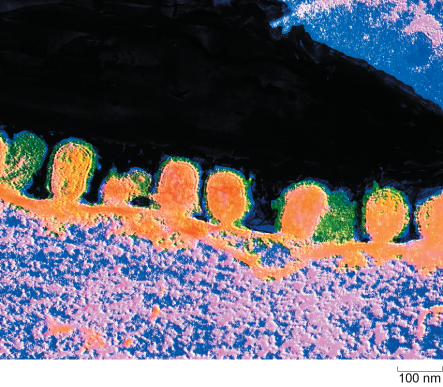
Influenza virus budding from the surface of an infected cell is shown in Figure 6.
From the description above, identify a process or element in the replication cycle which is characteristic of the virus, and which would not normally occur in a mammalian cell.
The replication of RNA on an RNA template with the production of double-stranded RNA would never normally occur in a mammalian cell. Double stranded RNA is therefore a signature of a viral infection. Significantly, cells have a way of detecting the presence of dsRNA, and this activates interferons: molecules involved in limiting viral replication.
2.5 Cellular pathology of influenza infection
Flu viruses can infect a number of different cell types from different species. This phenomenon is partly because the cellular glycoproteins which are recognised by viral haemagglutinin are widely distributed in the infectious agent.
What is the term for the property of viruses that allows them to only replicate in particular cell types?
This property is viral tropism. Hence we can say that flu viruses have a broad tropism.
A second reason why the virus can infect a variety of cell types is that the replication strategy of flu is relatively simple: ‘infect the cell, replicate as quickly as possible and then get out again’. This is the cytopathic effect of the virus. Cell death caused directly by the virus can be distinguished from cell death caused by the actions of the immune system as it eliminates infected cells.
The effects of cell death
Cell death impairs the function of an infected organ and often induces inflammation, a process that brings white cells (leukocytes) and molecules of the immune system to the site of infection. In the first instance, the leukocytes are involved in limiting the spread of infection; later they become involved in combating the infection, and in the final phase they clear cellular debris so that the tissue can repair or regenerate.
The symptoms of flu experienced by an infected person are partly due to the cytopathic effect of the virus, partly due to inflammation and partly a result of the innate immune response against the virus. The severity of the disease largely depends on the rate at which these processes occur.
- In most instances, the immune response develops sufficiently quickly to control the infection and patients recover.
- If viral replication and damage outstrip the development of the immune response then a fatal infection can occur.
In severe flu infections, the lungs may fill with fluid as the epithelium lining the alveoli (air sacs) is damaged by the virus. The fluid is ideal for the growth of bacteria, and this can lead to a bacterial pneumonia, in which the lungs become infected with one or more types of bacteria such as Haemophilus influenzae. Damage to cells lining blood vessels can cause local bleeding into the tissues, and this form of ‘fulminating disease’ was regularly seen in post-mortem lung tissues of people who died in the 1918 pandemic.
3 Patterns of disease
In humans, pigs and horses, flu viruses circulate through populations at regular intervals. The disease is endemic in tropical regions for all of these host groups (i.e. it is continually present in the community). In temperate latitudes, infections are usually seasonal or epidemic, with the greatest numbers occurring in the winter months (Figure 7). Epidemics also occur sporadically in sea mammals and poultry, and in these species high mortality is typical.
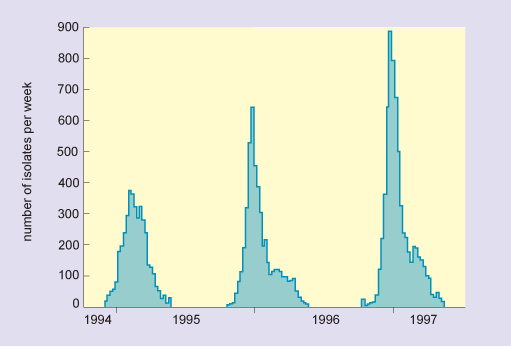
In most years, flu in humans affects a minority of the population, the disease course is not very severe and the level of mortality is not great. In such years the influenza virus is slightly different from the previous year due to antigenic drift, which results in the accumulation of genetic mutations that cause the molecules present on the surface of the virus change progressively. In this scenario, the virus is not significantly different from the previous year so that the host’s immune system can more easily mount an effective response than it could to a completely new strain.
However, at irregular intervals the virus undergoes an antigenic shift. This process only occurs in influenza A viruses, typically every 10–30 years, and it is associated with severe pandemics, serious disease and high mortality (see Box 2).
In Section 2.3 you read that strains of influenza are differentiated and designated using a simple system of numbers and letters that depend on their surface antigens. More commonly, however, strains responsible for pandemics are often given a common name according to the area in the world from which they were thought to originate, or the species they mainly affected before becoming transferred to humans (see Table 2). Evidence suggests, however, that in the twentieth century the major flu pandemics all originated in China, with the exception of the 1918 pandemic, which first occurred in the USA.
| Year | Designation | Common name |
|---|---|---|
| 1900 | H3N8 | (none) |
| 1918 | H1N1 | Spanish flu |
| 1957 | H2N2 | Asian flu |
| 1968 | H3N2 | Hong Kong flu |
| 1977 | H1N1 | Russian flu |
| 1997 | H5N1 | Avian flu |
Box 2 The rise of H5N1 – an example of antigenic shift
In 1997, a new strain of influenza A, H5N1, was identified in Hong Kong. The strain was rife in chickens and a few hundred people had become infected. Mortality in these individuals was very high, (6 of 18 died), and so there was serious concern that it marked the beginning of a new pandemic. The authorities in Hong Kong responded by a mass cull of poultry in the region and about 1.5 million chickens were slaughtered. H5N1 did not spread easily from person to person and no further cases were reported in people following the slaughter.
Whether the H5N1 outbreak was an isolated incident of a strain spreading from chicken to humans, or whether it was the start of a major pandemic which was nipped in the bud, cannot be known. Subsequent analysis showed that the high virulence of the new strain could partly be related to the new variant of haemagglutinin (H5), and partly to a more efficient viral polymerase. This outbreak clearly demonstrates the way in which bird influenza can act as a source of new viral strains, and shows that such new strains may be very dangerous to humans.
Since the discovery of the influenza virus in the 1930s it has been possible to isolate and accurately identify each of the epidemic strains, but, as earlier strains of virus have now died out, it has been necessary to infer their identity by examining the antibodies in the serum of affected people.
Antibodies and the ability of the immune system to respond to a strain of flu are much more persistent than the virus itself. It is thus possible to analyse antibodies to determine which types of haemagglutinin and neuraminidase they recognise long after the virus itself has gone. One can then deduce which type of influenza virus that person contracted earlier in their life (as explained in Section 5.2).
3.1 Tracking the emergence of new strains
Influenza is one of several diseases monitored by the WHO Global Alert and Response (GAR) network (WHO, 2011a), comprising 110 ‘sentinel’ laboratories in 82 countries. The organisation’s surveillance and monitoring of the disease then forms part of their Global Influenza Programme (GIP), and they use data gathered from participating countries to:
- provide countries, areas and territories with information about influenza transmission in other parts of the world to allow national policy makers to better prepare for upcoming seasons
- provide data for decision making regarding recommendations for vaccination and treatment
- describe critical features of influenza epidemiology including risk groups, transmission characteristics, and impact
- monitor global trends in influenza transmission
- inform the selection of influenza strains for vaccine production (WHO, 2011b).
The influenza data from the sentinel laboratories is fed into a global surveillance programme, started by the WHO in 1996, called FluNet (WHO, 2011c), which is one of the tools that facilitates the actions described above.
Activity 1 Using FluNet
Use the link below to visit the WHO’s FluNet web page and locate and view the chart showing the global circulation of flu (in the section marked ‘View charts’) to find out which flu subtypes are currently the major ones in circulation in the human population.
At the time of writing (2011), Influenza A (H5N1) and influenza B (unknown lineage) are the two main types globally. FluNet also breaks this information down into geographic areas and countries so, if you wish, you can see what subtypes are circulating where you live.
Typically a flu vaccine contains material from the main influenza A strains and an influenza B strain, so that an immune response is induced against the most likely infections. Usually the scientists predict correctly and immunised people are effectively protected against the current strains (>90% protection). However, the prediction is occasionally incorrect, or a new strain develops during the time that the vaccine is being manufactured. In this case the vaccine generally provides poor protection.
What can you deduce about immunity against flu infection from the observations on vaccination above?
The immune response is strain-specific. If you are immunised against the wrong strain of flu, then the response is much less effective and you are more likely to contract the disease.
3.2 Immune responses to influenza
The immune system uses different types of immune defence against different types of pathogen. The responses against flu are typical of those which are mounted against an acute viral infection, but different from the responses against infection by bacteria, worms, fungi or protist parasites.
When confronted with an acute viral infection, the immune system has two major challenges:
- The virus replicates very rapidly, killing the cells it infects. Since a specific immune response takes several days to develop, the body must limit the spread of the virus until the immune defences can come into play.
- Viruses replicate inside cells of the body, but they spread throughout the host in the blood and tissue fluids. Therefore, the immune defences must recognise infected cells (intracellular virus) and destroy them. But the immune system must also recognise and eradicate free virus in the tissue fluids (extracellular virus) in order to prevent the virus from infecting new cells.
The kinds of immune defence that the body deploys against flu are briefly considered in the next section.
3.2.1 Summary of the response
How does the body act quickly to limit viral spread?
When a virus infects a cell of the body, the molecular machinery for protein synthesis within the cell is usurped as the virus starts to produce its own nucleic acids and proteins. The cell detects the flu dsRNA and other viral molecules and releases interferons, which bind to receptors on neighbouring cells and cause them to synthesise antiviral proteins. If a virus infects such cells they resist viral replication, so fewer viruses are produced and viral spread is delayed.
Also, in the earliest stages of a virus infection the molecules on the cell surface change. Cells lose molecules that identify them as normal ‘self’ cells. At the same time they acquire new molecules encoded by the virus. A group of large, granular lymphocytes recognise these changes and are able to kill the infected cell. This function is called ‘natural killer’ cell action and the lymphocytes that carry it out are termed NK cells.
Non-adaptive and adaptive responses
The actions of both interferons and NK cells in combating infection by influenza occur early in an immune response, and are not specific for the flu virus. These defences occur in response to many different kinds of viral infection, and they are part of our natural, or non-adaptive, immune responses.
Note that immunologists use the term non-adaptive to indicate a type of response that does not improve or adapt with each subsequent infection. This is quite different to its use in evolutionary biology, where it means ‘not advantageous’. Such non-adaptive immune responses slow the spread of an infection so that specific, or adaptive, immune defences can come into play.
The key features of an adaptive immune response are specificity and memory. The immune response is specific to a particular pathogen, and the immune system appears to ‘remember’ the infection, so that if it occurs again the immune response is much more powerful and rapid. Because an immune response is highly specific to a particular pathogen it often means that a response against one strain of virus is ineffective against another – if a virus mutates then the lymphocytes that mediate adaptive immunity are unable to recognise the new strain.
There are two principal arms of the adaptive immune system, mediated by different populations of lymphocytes. One group, called T-lymphocytes, or T cells (which develop in the thymus gland, overlying the heart), recognises antigen fragments associated with cells of the body, including cells which have become infected. A set of cytotoxic T cells (Tc) specifically recognises cells which have become infected and will go on to kill them. In this sense they act in a similar way to NK cells. However they differ from NK cells in that Tc cells are specific for one antigen or infectious agent, whereas NK cells are non-specific.
The second group of lymphocytes are B cells (that differentiate in the bone marrow), which synthesise antibodies that recognise intact antigens, either in body fluids or on the surface of other cells. Activated B cells progress to produce a secreted form of their own surface antibody. Antibodies that recognise the free virus act to target it for uptake and destruction by phagocytic cells.
Therefore the T cells and NK cells deal with the intracellular phase of the viral infection, while the B cells and antibodies recognise and deal with the extracellular virus.
The two reactions described above are illustrated in Figure 8.
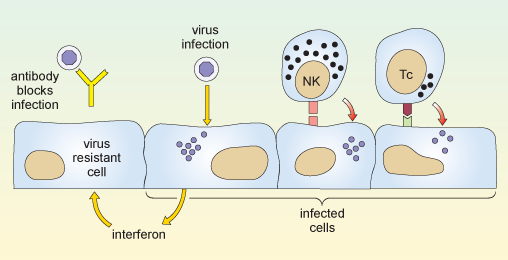
You might ask why it takes the adaptive immune response so long to get going. The answer is that the number of T cells and B cells that recognise any specific pathogen is relatively small, so first the lymphocytes which specifically recognise the virus must divide so that there are sufficient to mount an effective immune response. This mechanism is fundamental to all adaptive immune responses.
Activity 2 Influenza mini-lecture
Some of the themes that we have discussed up to now are presented in Video 1: a mini-lecture on influenza by David Male of The Open University. Watch the videos and then attempt the questions below
Transcript: Video 1 Influenza mini-lecture.
Influenza is a viral disease which generally starts with an infection of the upper respiratory tract and develops with systemic symptoms including fever and lassitude over the course of 1–2 weeks. Although it is debilitating, most people make a full recovery. However it may be fatal for older people and those with weaker immune systems, particularly if the virus disposes them to concurrent bacterial infections such as pneumonia.
The disease is caused by a group of myxoviruses, influenza A, B and C, although the most serious infections are caused by influenza A, and the following discussion refers to influenza A. The virus is seen in this transmission electron micrograph with proteins projecting from the viral envelope. Very many different variants of influenza have been identified since the virus was first discovered in 1935, and the variants are responsible for the different infections which occur in successive years. This is the reason that we can suffer from flu several times during our lives – in effect we become infected on each occasion with a different strain of virus, which our immune system has not encountered before and to which we are not immune.
Look at the overall structure of influenza A. The myxoviruses are all enveloped, that is to say that they have an outer membrane or envelope, which has been derived from the plasma membrane of an infected host cell. Two major viral proteins are present in the envelope, namely the haemagglutinin and the neuraminidase. Neuraminidase is an enzyme which cleaves sialic acid residues on glycoproteins and this protein performs an important function, in allowing the virus to bud off from the infected cell and spread through the body. The haemagglutinin is also essential for viral infection. It binds to carbohydrate groups present on glycophorins, molecules which occur on the surface of cells of the body. Binding of haemagglutinin to the surface of host cells is the first step in infection. Since the glycophorins are widely distributed on different cell types, influenza is able to cause widespread infection. As we shall see later, the antibody response against the haemagglutinin of the virus is critical in protecting us against on-going infection, although antibodies to the neuraminidase can also contribute to immunity. Within the viral envelope, the viral capsid is formed from the ‘M-protein’, which contains the virus’ genetic material, RNA, nucleoproteins, and a number of enzymes needed for replication.
The viral genome consists of 8 separate strands of RNA. Because the genome is fragmented in this way, it means that the different strains of virus can re-assort their genes relatively easily, and this is an important source of new viral strains.
Look now at the epidemic pattern of influenza over a number of years. The graph shows the number of isolates of different strains of influenza, in laboratories in the USA between 1995 and 1997. You can see that in temperate latitudes the infection rates follow a seasonal pattern, with more people developing the disease in the winter months. Examination of the structure of the haemagglutinin in successive years shows that minor mutations occur in the primary structure between the virus strains prevalent in each year. Although these do not affect the ability of the haemagglutinin to bind to host cells, the change is sufficient to prevent antibodies specific for a previous strain from binding to the new strain’s haemagglutinin. In effect, last year’s antibodies are unable to protect us from this year’s strain of flu.
The structure of the viral haemagglutinin can be seen in this model, which shows the backbone of the chain of amino acids. We will look at the way in which a neutralising antibody binds to an epitope on the haemagglutinin. The epitope is formed by amino acid residues in the loops at the exposed part of the molecule. Three loops which contribute residues to the epitope are highlighted here by space-filling the residues. Two domains of the heavy chain of the neutralising antibody are shown in yellow. The constant and variable domains are clearly visible, and the three hypervariable loops which contribute to the binding site are picked out in colour. You can see how the epitope and the heavy chain are complementary in shape. To complete the picture, we will add the light chain to the model. In this case, it is clear that the light chain contributes very little to the antibody combining site. When the space-filling model is completed, you can see that the residues forming the epitope are buried in the centre of the binding site and it is precisely these residues which are most likely to mutate between one viral strain and another. This progressive but limited change in the structure of the haemagglutinin is called genetic drift. Occasionally, perhaps once every 10–20 years, a major new strain of influenza appears which is radically different from those of previous years. Such a change is called genetic shift. The appearance of such new strains is associated with a worldwide serious epidemic of flu, called a pandemic. For example the pandemic strain of ‘Spanish flu’ which developed in 1918, had a different haemagglutinin and neuraminidase from the previously dominant strain, and this outbreak is thought to have caused the deaths of 20 million people world-wide. The picture shows a ward at the time. This epidemic particularly affected young fit people. One doctor wrote ‘it is only a matter of hours until death comes. It is horrible.’
The origin of new pandemic strains has been much debated, but it appears most likely that a human strain of flu exchanges genetic material with an animal strain of flu for example from ducks, or pigs. Such a reassortment of genes could occur if two different flu viruses simultaneously infect the same cell, and produce new viruses containing some gene segments from each type – remember that the flu virus has a segmented genome which allows this to occur.
The major pandemic strains are distinguished according to which haemagglutinin they have and which neuraminidase. So, for example in 1957 the dominant strain H1, N1 changed both its haemagglutinin and neuraminidase, and the new dominant strain H2N2 persisted until 1968. At present, (that is, in 2001) there are two dominant strains of influenza A in circulation H1N1 and H3N2.
One of the problems of producing vaccine for flu is that we do not know what next year’s major strain will be, and there is only a limited amount of time available before an epidemic spreads. The map shows the way in which the epidemic of Asian flu spread in 1957 from its origin in China in February, through South-East Asia by April and from there to all parts of the world by the end of the year. The figures on the map indicate the months in which the virus was isolated in different areas.
Nowadays, laboratories throughout the world track the appearance of new variants, and aim to identify the current circulating strains and any potentially new pandemic strains. Having decided the composition of the vaccine, there are just a few months to prepare it for the next flu season. Have a look at the current vaccine, it contains examples of the two major strains of influenza A H1N1 and H3N2 which are circulating and vaccine for the main current strain of influenza B. As there is insufficient time and resource to produce vaccine for everyone, the vaccine is usually recommended for older people and high risk groups, such as health professionals.
In addition to the antibody response, cytotoxic T cells are important in clearing virally-infected cells. The cytotoxic T cells recognise peptide fragments of several of the viral proteins, including the internal proteins such as the M-protein nucleoproteins and polymerases which are genetically stable. The bar chart shows the ability of lymphocytes from a single donor to kill cells which have been transfected with a single flu antigen. This is a measure of the prevalence of cytotoxic T cells for each of the proteins. Most of the cytotoxicity is directed against internal proteins shown on the green bars. Clones of cytotoxic cells against internal proteins usually recognise several of the major strains of flu. In contrast, those which recognise the external proteins are often, but not always strain-specific.
Even when a cytotoxic T cell recognises the same antigen as an antibody, it usually recognises a different portion. For example cytotoxic T cells specific for the haemagglutinin often recognise internal fragments of the antigen rather than the external epitopes recognised by antibodies. Moreover, since cytotoxic T cells recognise antigen presented by MHC molecules, and since MHC molecules are different in each individual, the T cells in each individual recognise different parts of the antigen. These bar charts show the response of two individuals against peptides from the haemagglutinin. T cells from the first person respond to four different regions of the molecule, with highly antigenic peptides centred on residues 100, 180, 300 and 400. T cells from the second individual recognise different regions of the haemagglutinin.
There is some evidence that individuals with specific MHC haplotypes may be more efficient than others at recognising and destroying influenza-infected cells.
In summary, influenza A gives us an example of extreme genetic variability, where successive dominant strains of flu emerge. The new strains are not susceptible to control by antibodies in the host population, and so individuals may suffer from repeated infections. Indeed the general immunity in the host population provides the selective pressure for the emergence of the new strains.
- Draw a labelled diagram of the structure of influenza A.
- How do pandemic strains of influenza A come about?
Answer
Question 1
Your diagram should look like Figure 4.
Question 2
Pandemic strains of influenza A normally arise by simultaneous infection of a non-human host (typically poultry or pigs) with two or more strains of influenza A. Reassortment of the eight viral segments from each virus allows the generation of a new hybrid virus type, with a completely novel surface structure that has never been seen before by a host immune system (antigenic shift).
4 Antiviral treatments
Two classes of antiviral drugs are used to combat influenza: neuraminidase inhibitors and M2 protein inhibitors.
Why are antibiotics not used to combat influenza?
Influenza is a virus. Antibiotics only work against bacteria.
Neuraminidase inhibitors
Recall from Section 2.1 that neuraminidase is an enzyme that is present on the virus envelope and cleaves sialic acid groups found in the polysaccharide coating of many cells (especially the mucus coating of the respiratory tract). Neuraminidase is used to clear a path for the virus to a host cell and facilitates the shedding of virions from an infected cell. Inhibition of neuraminidase therefore helps prevent the spread of virus within a host and its shedding to infect other hosts.
The two main neuraminidase inhibitors currently in clinical use are zanamivir (trade name Relenza) and oseltamivir (trade name Tamiflu). These are effective against influenza A and B, but not influenza C which exhibits a different type of neuraminidase activity that only cleaves 9-O-acetylated sialic acid.
M2 inhibitors
Recall from Table 1 that the influenza M2 protein forms a pore that allows protons into the capsid, acidifying the interior and facilitating uncoating.
Drugs such as amantadine (trade name Symmetrel) and rimantadine (trade name Flumadine) block this pore, preventing uncoating and infection. However, their indiscriminate use in ‘over-the-counter’ cold remedies and farmed poultry has allowed many strains of influenza to develop resistance. Influenza B has a different type of M2 protein which is largely unaffected by these drugs.
5 Diagnosis of influenza
Many diseases produce symptoms similar to those of influenza; in fact, ‘flu-like’ is a term that is frequently used to describe several different illnesses. Since influenza spreads rapidly by airborne transmission and is a life-threatening condition in certain vulnerable groups, it is important that cases of the disease are identified as quickly as possible, so that preventative measures may be taken.
Most viral infections are not treated, although antiviral drugs such as zanamivir are used for potentially life-threatening cases or where the risk of transmission is high (as occurs during a pandemic).
Which sites in the body should be sampled for diagnosis?
The influenza virus infects the respiratory tract and is spread by coughing and sneezing, so specimens should be taken from the nose, throat or trachea.
In practice, the best specimens are nasal aspirates or washes, but swabs of the nose or throat may be used if they are taken vigorously enough to obtain cells. Ideally, samples should be taken within three days of the onset of illness, and all specimens need to be preserved in a transport medium and kept chilled until they reach the clinical microbiology laboratory.
5.1 Initial identification of influenza infection
Oral swabs or nasal aspirates are initially screened for the presence of a variety of respiratory viruses. This is done by extracting RNA from the sample and subjecting it to a reverse-transcription polymerase chain reaction (RT-PCR) as described below:
- Initially the RNA sample is reverse transcribed into complementary DNA (cDNA), using a commercially-available reverse transcriptase enzyme.
- The cDNA is then used in a standard PCR reaction to detect and amplify a short sequence of nucleotides specific to the virus. Multiple DNA sequences, each specific for a different type of virus, can be amplified in the same reaction, provided that these sequences are of different lengths.
- Each of the different amplified sequences is separated from the others when the entire sample is subjected to gel electrophoresis (an analytical technique in which molecules of different sizes move at various rates through a gel support in an applied electric field, thus making it possible to identify specific molecules.)
The polymerase chain reaction (PCR) technique is illustrated in Video 2.
Transcript: Video 2 PCR technique.
Polymerase chain reaction, or PCR, uses repeated cycles of heating and cooling to make many copies of a specific region of DNA. First, the temperature is raised to near boiling, causing the double-stranded DNA to separate, or denature, into single strands. When the temperature is decreased, short DNA sequences known as primers bind, or anneal, to complementary matches on the target DNA sequence. The primers bracket the target sequence to be copied. At a slightly higher temperature, the enzyme Taq polymerase, shown here in blue, binds to the primed sequences and adds nucleotides to extend the second strand. This completes the first cycle.
In subsequent cycles, the process of denaturing, annealing and extending are repeated to make additional DNA copies.
After three cycles, the target sequence defined by the primers begins to accumulate.
After 30 cycles, as many as a billion copies of the target sequence are produced from a single starting molecule.
Typically, nucleotide sequences specific to five types of virus are searched for in each sample: influenza A, influenza B, respiratory syncytial virus (Baltimore group V, (–)ssRNA virus, and a major cause of respiratory illness in young children), adenoviruses and enteroviruses. Those samples that test positive for influenza in the RT-PCR reaction are inoculated into cells in culture. Sufficient virus for a limited number of tests can be produced from such cultures within 24 hours, but they are often maintained for up to a week.
5.2 Determining the subtype of influenza
Immunofluorescence
Confirmation of a case of influenza is usually achieved by performing tests on some of the inoculated cultured cells using reference fluorescent-labelled antisera provided by the WHO. A reference antiserum is a sample serum known to contain antibodies specific for the molecule to be assayed (in this case, haemagglutinin or neuraminidase). These antisera are prepared using purified haemagglutinin and neuraminidase and are monospecific, each antibody reacting only with one epitope e.g. H1 or H3.
For the test, an antibody is added to a sample of inoculated cells. Following a wash step, if the antibody remains bound to the cells then they fluoresce under appropriate illumination, indicating the presence of viral antigen on the cell surface. This diagnostic technique can identify influenza virus on infected cells in as little as 15 minutes. A positive result not only confirms the RT-PCR data, but gives additional information on the subtype of the virus.
Further PCR analyses
Standardised RT-PCR protocols exist to look for the presence of different haemagglutinin and neuraminidase subtypes, chiefly H1, H3, H5, N1 and N2. (Poddar, 2002). If the PCR analysis indicates a dangerous strain of influenza A e.g. H5N1, then it is instantly sent to a WHO reference laboratory for further tests.
Haemagglutination assays
Influenza has haemagglutinins protruding from its viral envelope, which it uses to attach to host cells prior to entry. These substances form the basis of a haemagglutination assay, in which viral haemagglutinins bind and cross-link (agglutinate) red blood cells added to a test well, causing them to sink to the bottom of the solution as a mat of cells. If agglutination does not occur, then the red blood cells are instead free to roll down the curved sides of the tube to form a tight pellet.
A related test called a haemagglutination-inhibition assay (HAI), incorporates antibodies against different subtypes of viral haemagglutinin. The antibodies bind and mask the viral haemagglutinin, preventing it from attaching to and cross-linking red blood cells.
A HAI assay can be set up in one of two ways: either a known reference antibody is added to an unknown virus sample, or known reference viral haemagglutinin is added to a sample of patient serum containing antibodies against influenza. This second version of the HAI assay can therefore be used long after the infection has passed, when virions are no longer present.
Haemagglutination and HAI assays have the advantage that they are simple to perform and require relatively cheap equipment and reagents. However, they can be prone to false positive or false negative results, if the sample contains non-specific inhibitors of haemagglutination (preventing agglutination) or naturally occurring agglutinins of red blood cells (causing agglutination).
If a confirmed influenza A isolate reacts weakly or not at all in HAI then this indicates an unknown variant of influenza A and the sample is immediately sent to a WHO reference laboratory for further tests.
Neuraminidase inhibition assay
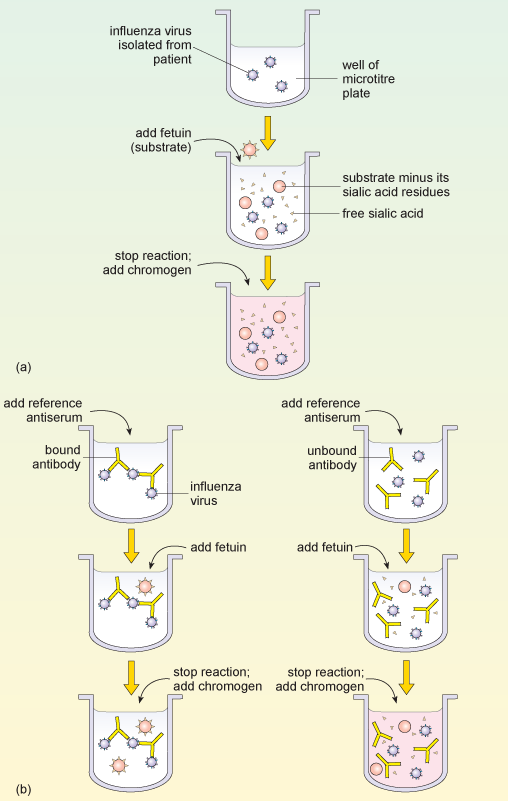
Typing influenza isolates in terms of their neuraminidase makes use of the enzyme activity of this glycoprotein. The neuraminidase inhibition assay is performed in two parts. The first part determines the amount of neuraminidase activity in a patient influenza sample, as outlined in Figure 9a. A substrate (called fetuin) that is rich in sialic acid residues is added to a sample of the influenza virus, and the viral neuraminidase enzyme cleaves the substrate to produce free sialic acid.
Addition of a substance that inactivates the neuraminidase stops the reaction, and a chromogen (a colourless compound that reacts to produce a coloured end-product) that turns pink in the presence of free sialic acid is added. The intensity of the pink colour is proportional to the amount of free sialic acid and can be measured using a spectrophotometer.
This assay of neuraminidase activity allows the appropriate amount of virus sample to be determined, and this quantity is then used in the second part of the assay. If too much or too little virus is used, the resulting changes, and therefore the neuraminidase, may be undetectable.
In the second part of the assay (Figure 9b), viral samples from the patient are incubated with anti-neuraminidase reference antisera. Each of the reference antisera used for this test has antibodies that bind one particular neuraminidase variant, e.g. N1 or N2.
How can these antisera be used to type the neuraminidase variant?
If the antibodies bind the neuraminidase in the patient’s sample they inhibit its activity. This means that the patient’s neuraminidase cannot cleave the sialic acid from its test substrate fetuin, so no colour change will occur when the chromogen is added. Conversely, if the antibodies in the reference antiserum do not bind the neuraminidase in the patient’s sample, then the enzyme will remain uninhibited and the pink colour will be produced as before.
6 Conclusion
As you reach the end of this free course you should consider some of the important points that the study of flu raises.
- A single pathogen can produce different types of disease in different people. Genetic variation in a pathogen can also affect the type of disease it produces. To understand this we need to know something of the genetic and social differences in the host population, and of the diversity of the pathogen.
- The symptoms of a particular disease may be produced by different pathogens or by a combination of pathogens. To understand this requires some knowledge of pathology and cell biology.
- Some diseases, such as flu, affect humans and several other animal species, whereas others are more selective in their host range. The basic biology of different pathogens underlies these differences.
- Flu is a disease that can be contracted several times during a lifetime, but many other infectious diseases are only ever contracted once. To understand this we need to look at how the immune system reacts to different pathogens, and how responses vary depending on the pathogen.
- Outbreaks of flu occur regularly, but some epidemics are much more serious than others. This requires an understanding of aspects of virology, immunology, evolutionary biology and epidemiology.
7 Questions for the course
The following questions allow you to assess your understanding of the content of this course. Each one relates to one or more of the intended learning outcomes of the study.
If you are unable to answer a question, or do not understand the answer given, then reread the relevant section(s) of the course and try the question again.
Question 1
(This question relates to case study learning outcome (LO) 2.)
Why would Robert Koch have been unable to demonstrate that influenza viruses cause the disease influenza, according to his own postulates?
Answer
Koch’s second postulate states that the pathogen can be isolated in pure culture on artificial media. Viruses can only multiply within host cells, so Koch would have been unable to isolate the virus using artificial media. (Much later, eggs and live cells in tissue culture came to be used for growing flu virus, but this was long after Koch’s death, and they do not strictly conform to the original postulate.)
Question 2
(This question relates to LO2.)
List the various structural components of an influenza A virus and note where each of these elements is synthesised within an infected cell.
Answer
Viral RNA is synthesised in the nucleus of the infected cell. The M-protein and other internal proteins are synthesised on ribosomes in the cytoplasm. The capsid is then assembled in the nucleus. The haemagglutinin and neuraminidase are synthesised on ribosomes on the endoplasmic reticulum. The envelope is derived from the host cell’s own plasma membrane.
Question 3
(This question relates to LO2 and LO4.)
It is very uncommon for a strain of influenza that infects other animals to infect people; nevertheless such strains are very important for human disease. Why is this?
Answer
Animal strains of influenza act as a reservoir of genes that may recombine with human influenza viruses to produce new strains that can spread rapidly in humans. Such pandemic strains frequently produce serious diseases with high mortality.
Question 4
(This question relates to LO2 and LO3.)
Which immune defences are able to recognise and destroy virally-infected host cells?
Answer
Cytotoxic T cells and NK cells are able to recognise and destroy virally-infected host cells.
Question 5
(This question relates to LO4 and LO5.)
Why do most people suffer from influenza several times in their lives?
Answer
The virus mutates regularly (antigenic drift); also new strains are occasionally generated by recombination (antigenic shift). Since the immune response is generally specific for a particular strain of virus, new strains are not susceptible to immune defences which have developed against earlier strains.
Glossary
- adaptive
- The adaptive immune defence refers to the tailoring of an immune response to the particular foreign invader. It involves differentiating self from non self and involves B cells and T cells (lymphocytes). A key feature of the adaptive immune system is memory. Repeat infections by the same virus are met immediately with a strong and specific response.
- antigenic drift
- Minor changes in antigens that occur as a pathogen mutates.
- antigenic shift
- Major changes in the antigens of a pathogen that result from reassortment of genes.
- cytopathic
- Description of the damage caused to host cells or tissues by a pathogen, often a virus.
- gel electrophoresis
- The separation of molecules (proteins or nucleic acids) in an electric field as a function of their size and charge.
- genetic reassortment
- The mixing of the genetic material of two species into new combinations in different individuals. Especially used to describe when two or more similar viruses, infecting the same cell, exchange genetic material to produce a new virus.
- genome
- The complete set of genes that an organism contains.
- inflammation
- A series of reactions, which bring cells and molecules of the immune system to sites of infection or damage. This appears as an increase in blood supply, increased vascular permeability and increased transendothelial migration of leukocytes.
- non-adaptive
- The non-adaptive immune defence (sometimes called the ‘innate’ or ‘humoral’ response) is a non-specific response and is generally the first line of immune defence, being active even before infection begins. Receptors on host cells detect uniquely viral components, such as double stranded RNA and viral glycoproteins, and trigger the release of cytokines.
- T cells
- Lymphocytes that differentiate primarily in the thymus and are central to the control and development of immune responses. The principle subgroups are cytotoxic T cells (Tc) and T helper cells (Th).
References
Further reading
Acknowledgements
This course was written by Jon Golding and Hilary MacQueen.
Except for third party materials and otherwise stated (see terms and conditions), this content is made available under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 Licence.
Course image: thierry ehrmann in Flickr made available under Creative Commons Attribution 2.0 Licence.
The material acknowledged below is Proprietary and used under licence (not subject to Creative Commons Licence). Grateful acknowledgement is made to the following sources for permission to reproduce material in this course:
Figure 1: Prescott, L., Harley, J., and Klein, D. (1999) Microbiology, 4th ed. Copyright © The McGraw-Hill Companies
Figure 3: Noymer, A., and Garenne, M. (2000) ‘The 1918 influenza epidemic’s effects on sex differentials in mortality in the United States’, Population and Development Review, Vol 26 (3) 2000, The Population Council
Figure 6: CNRI/Science Photo Library;
Figure 7: Bammer, T. L. et al. (April 28, 2000) ‘Influenza virus isolates’ reported from WHO, Surveillance for Influenza – United States, Centers for Disease Control and Prevention
Video 1: Immunology Interactive (Male, Brostoff and Roitt) copyright the authors, reproduced by permission of David Male
Video 2: with kind permission from the Howard Hughes Medical Institute.
Every effort has been made to contact copyright owners. If any have been inadvertently overlooked, the publishers will be pleased to make the necessary arrangements at the first opportunity.
Don't miss out:
If reading this text has inspired you to learn more, you may be interested in joining the millions of people who discover our free learning resources and qualifications by visiting The Open University - www.open.edu/ openlearn/ free-courses