Vaccination
Use 'Print preview' to check the number of pages and printer settings.
Print functionality varies between browsers.
Printable page generated Thursday, 2 May 2024, 10:40 PM
Vaccination
Introduction
The course begins with the early history of smallpox – the first infectious disease to be eradicated by a vaccination programme. At the end of section 1, we ask you to read an article on the history of smallpox, then, before continuing further with this course, you should turn to the case study on polio, where we discuss the prospects for making this the second infectious disease to be eradicated by vaccination. At the end of section 5 you will study the mini-lecture on vaccination. You will conclude your study of this chapter by conducting some internet research on the progress of vaccination programmes.
This OpenLearn course provides a sample of Level 3 study in Science.
Learning outcomes
After studying this course, you should be able to:
define and use, or recognise definitions and applications of, each of the terms in bold in the course
use examples from the history of vaccination to illustrate the conduct and outcomes of vaccine strategies to control infectious diseases
discuss the principle strategies available for developing a vaccine and explain the significance of critical antigens, immunogens and adjuvants in developing effective vaccines
identify examples of infectious diseases for which effective vaccines are available and some for which they are not. Explain why it has been scientifically difficult or commercially unprofitable to develop vaccines against certain infectious diseases, and why others have been amenable to control by vaccination
discuss the prospects for developing a vaccine against a named infectious disease, given information on its biology and epidemiology, and on the immune response in human hosts.
1 Smallpox and the history of vaccination
1.1 The smallpox virus
Undoubtedly, one of the great success stories of modern medicine has been in the field of vaccination against infectious diseases. There is no more compelling example than smallpox. It is hard now to imagine the impact of smallpox, which killed 10–50% (sometimes more) of the people it infected and wiped out whole communities. In the nineteenth century, the English parliamentary historian Thomas Macaulay graphically described its effects:
Smallpox was always present, filling the churchyard with corpses, tormenting with constant fear all whom it had not yet stricken, leaving on those whose lives it spared the hideous traces of its power, turning the babe into a changeling at which the mother shuddered, and making the eyes and cheeks of the betrothed maiden objects of horror to the lover.
(From The History of England from the Accession of James II by Thomas Macaulay, completed 1855)
Smallpox, caused by the variola virus, is thought to have originated in the first agricultural settlements in North Africa and was one of the greatest scourges of humanity for at least 10000 years. It holds a special place in the history of immunology and infectious disease as the first disease for which an effective vaccine was developed and the first to be eradicated globally (confirmed by the WHO in 1980).
1.2 Variolation
By the seventeenth century, the observation that immunity from severe smallpox followed a mild episode of the disease had led to the practice of ‘variolation’ in China and the Ottoman Empire. Variolation was so named because material was taken from dried scabs or pustules (the Latin varus, 'marks on the skin’) and used deliberately to infect healthy recipients. Infected material could be applied to an area of scarified skin or could be introduced into the nose as a dust. The material was taken from people with a milder form of the disease (possibly caused by variola minor virus). Although there was no understanding of either the immune system or infectious agents at that time, the effect was to expose the recipient to a supposedly ‘weaker’ strain of the virus, which elicited a protective immune response against subsequent infection with a more virulent strain.
Activity 1
What key features of the immune response does the practice of variolation demonstrate?
Answer
It shows the specificity of the immune response (variolation with smallpox material protected recipients specifically against this disease) and the existence of immunological memory (it resulted in long-lasting protection).
Variolation was usually followed by a fever and the treated area of skin developed an acute inflammatory response with the characteristic ‘pox’ lesions. Although recovery usually ensued, 2–3 per cent of people could be killed by the smallpox infection that resulted from the procedure.
Activity 2
Why would a ‘medical’ procedure with a 2–3 per cent fatality rate be tolerated?
Answer
The risk of fatality from variolation was still much less than that of contracting a fatal smallpox infection.
Variolation was introduced into Europe early in the eighteenth century (you will learn more about this period when you read the article at the end of this section), where it gradually became known as inoculation (from the Latin for ‘to graft or implant’).
1.3 Edward Jenner and vaccination with cowpox

Vaccination originally meant deliberate infection with the cowpox virus (vaccinia), which is responsible for a relatively benign infection on the udders of cows and can be transferred to people, where it usually causes pustules on the hands. However, serious complications can ensue in a minority of cases. Vaccination with cowpox developed from an experiment carried out by an English country doctor, Edward Jenner (Figure 1). He had heard the common folklore that milkmaids who became infected with cowpox appeared to be protected from smallpox, and it had been previously reported that people who had recovered from cowpox did not develop the usual skin reaction to variolation. In 1796, Jenner deliberately infected an eight-year-old boy with the cowpox virus and repeated the experiment on ten others in the next two years. He confirmed that these vaccinated subjects did not respond to smallpox variolation and, despite initial resistance (Figure 2), his work ushered in the era of protective immunisation. In honour of Jenner, the term vaccination became widely used for any procedure in which the aim is to produce or enhance immunity to an infectious agent. In this course, we follow this tradition, but note that vaccination and immunisation are equivalent terms in current usage.

Activity 3
The type of vaccination developed by Jenner used one kind of pox virus to produce immunity against another. How can an immune response against one antigen or pathogen be effective against another? Does this not go against the idea that immune responses are ‘specific’?
Answer
An antibody that binds to an epitope (a particular molecular shape) on one antigen will also bind to another antigen if it shares an identical epitope, or a very similar one. Two viruses may have sufficiently similar epitopes that an antibody raised against one will also bind to the other. Antibody specificity is not absolute.
Although it was the first to be discovered, this type of vaccination – using one pathogen to protect against another with which it cross-reacts – is quite unusual. Much more common is the use of killed pathogens, or a harmless variant of the pathogen, or one of its component antigens, to induce immunity without producing disease. We look at modern methods of vaccine production in Section 4.
1.4 Is smallpox still a threat?
Since the smallpox virus was declared eradicated ‘in the wild’ in 1980, stocks of virus have been held in secure laboratories in various parts of the world, with the expectation that they would eventually be destroyed. However, since 2001, the perceived threat of bioterrorism has led to debates about whether they should be retained as a vital resource for research into ways to combat a deliberate release of smallpox virus – assuming that samples have been (or could be) obtained by terrorist groups. Mathematical models of how smallpox might spread through a population have been constructed, and the effects of vaccinating people with vaccinia either before or after a theoretical exposure have also been modelled.
Vaccination programmes ended over 20 years ago, so even those individuals who were vaccinated as children are unlikely now to be protected against smallpox (i.e. almost everyone is susceptible), but a decision on whether to reintroduce smallpox vaccination is not straightforward. Serious complications of injecting people with vaccinia can be expected to occur in a minority of individuals – particularly those with immunodeficiency. In the 1960s, smallpox vaccination led to an estimated 1–3 deaths per million doses, but today's population now has very much larger numbers of people whose immune system is suppressed by HIV infection or medical treatment (e.g. for cancer), or following organ transplants. The vaccine contains ‘live’ vaccinia, so even if its recipients are selected to be in good health, they can pass the virus on to others who may be less able to withstand its pathological effects.
So the question of whether the risks of reintroducing smallpox vaccination outweigh the possible benefits depends on estimates of the potential risk of a bioterrorist attack. At the time of writing (2003) the threat is considered to be so low that vaccination has only been reintroduced in the UK and USA for health workers who would be in the ‘front line’ of any response to an outbreak.
1.5 Summary of Section 1
-
Smallpox, caused by the variola virus, was one of the great pandemic infectious diseases for more than 10,000 years, killing a high proportion of infected people and changing the course of history.
-
Variolation in the seventeenth and eighteenth centuries used material from ‘mild’ smallpox cases to infect healthy people, most of whom developed protective immunity, but there was a 2–3 per cent death rate.
-
In 1796 Edward Jenner began experiments that led to widespread vaccination with cowpox virus (vaccinia), which elicits antibodies that cross-react with variola and protect against smallpox. By 1980, vaccination had eradicated smallpox globally.
-
Since 2001, the costs and benefits of reintroducing smallpox vaccination have been debated in response to the possible threat of bioterrorism.
Find out more about polio by reading the Case Study below, where we discuss the biology and epidemiology of polio and reflect on the global vaccination programme, which aimed to eradicate it from the world by 2005. You should allow around three hours for this, including some time for exploration of WHO and UNICEF websites, which publish regularly updated information on the progress of the polio eradication campaign. There is also an optional visit to a website on the social history of polio epidemics in the USA in the twentieth century.
Click to view the Polio Case Study.
2 Active vaccines and passive immunisation
2.1 From passive to active
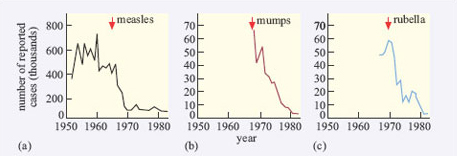
Since Jenner's pioneering discovery, many new vaccines have been developed (Table 1). The country in which a vaccine was first introduced is usually the one that developed it; France and the USA are among the most prominent: for example, rabies, plague and BCG vaccines were first used in France, polio vaccines were introduced first in the USA. There has often been a considerable time lag between dates of first use in the country in which a vaccine was pioneered and its adoption elsewhere (e.g. BCG vaccination was delayed in the UK until 1954), and some countries have never adopted particular vaccines. Some were immediately introduced in mass vaccination programmes (e.g. against polio), and others have only been used selectively to control outbreaks. Continuing research means that the ‘first’ vaccines against a particular infectious disease are superseded by more effective preparations. Some vaccines are now so effective that the infections they protect against are termed vaccine-preventable diseases – the WHO has placed the highest priority on achieving mass vaccination against diphtheria, whooping cough (pertussis), tetanus, measles, mumps, rubella, polio and TB.
The incidence of most of the diseases in Table 1 was declining in most countries in the world for some time before the introduction of the relevant vaccine, due to improvements in public health and living standards. However, the annual toll of mortality and morbidity was significantly greater than it is today and sharp declines in the incidence occurred when an effective vaccination programme was introduced, as Figure 3 and the Polio Case Study illustrate.
| Infectious disease | First use |
|---|---|
| smallpox | 1798 |
| rabies | 1885 |
| cholera | 1885 |
| tetanus antitoxin (passive) | 1890 |
| diphtheria antitoxin (passive) | 1893 |
| anthrax | 1891 |
| typhoid | 1896 |
| plague | 1897 |
| diphtheria | 1923 |
| tuberculosis (BCG) | 1923 |
| tetanus | 1924 |
| pertussis (whooping cough) | 1926 |
| tetanus | 1927 |
| yellow fever | 1935 |
| hepatitis A (passive) | 1945 |
| polio (IPV) | 1955 |
| polio (OPV) | 1962 |
| measles | 1963 |
| mumps | 1967 |
| meningitisA | 1969 |
| rubella | 1970 |
| haemophilus influenza | 1972 |
| viral influenza | 1976 |
| meningitis C (polysaccharide) | 1977 |
| hepatitis B | 1981 |
| hepatitis A | 1989 |
| varicella zoster (chickenpox) | 1995 |
| meningitis C (conjugate) | 1999 |
When people use the term vaccination, they nearly always mean active vaccination, i.e. immunising an individual with pathogen-specific antigens in order to induce a protective immune response against subsequent infection with that pathogen. The antigens in the vaccine elicit a primary immune response, which takes 7–14 days to reach its peak before subsiding. ‘Booster’ doses of the vaccine increase the protection against that infection by ensuring that it will be met by an enhanced secondary immune response.
Activity 5
Explain why the antibodies produced during the secondary response are more effective than those of the primary response.
Answer
There is a shorter lag time before pathogen-specific antibodies appear (typically 2–3 days); the overall level of antibodies is higher and they persist for longer; less IgM and more IgG is produced (due to class switching), which increases the protection in the blood stream; the affinity of the antibody binding sites for the target antigen is generally higher, so they bind more strongly.
In a small number of infectious diseases, active vaccines can also be given therapeutically to people who are already infected, to stimulate their immune system to eliminate the pathogens, or at least slow the progression of the disease. This approach is being tested extensively in trials of therapeutic vaccines against established HIV infection.
However, there is another type of procedure, termed passive immunisation, which is used therapeutically to treat particular infectious diseases after symptoms have developed, or for post-exposure prophylaxis. In passive immunisation, the recipient is directly injected with antibodies from another immune individual, who had either developed immunity following an infection or after immunisation with an active vaccine. The procedure is described as ‘passive’ because recipients do not manufacture the antibodies for themselves. Indeed, the presence of passively acquired antibodies may reduce the ability of recipients to manufacture their own antibodies (because of negative feedback controls operating in the immune system).
Activity 6
Under what circumstances would it be advantageous to give someone pre-prepared antibodies against an infectious agent?
Answer
Passive immunisation is particularly important in certain life-threatening infections where there are no anti-infective drugs or they act too slowly; active vaccination takes too long to stimulate a protective immune response.
The most important infections in which passive immunisation is used are listed in Table 2. The use of passive immunisation often preceded the introduction of an active vaccine against certain infections (see Table 1 earlier)
| Disease | Source of antibodies | Usage |
|---|---|---|
| tetanus | human or horse serum | after symptoms develop |
| diphtheria | human or horse serum | after symptoms develop |
| gangrene | horse serum | after exposure |
| botulism | horse serum | after exposure |
| hepatitis B | human serum | after exposure |
| rabies | human serum | after exposure (plus active vaccine) |
Activity 7
Explain why pathogen-specific antibodies are given immediately to people with suspected tetanus, diphtheria or botulism. How do they protect the patient?
Answer
All three bacterial diseases are caused by a potentially fatal exotoxin, and it is imperative to neutralise it in the blood as quickly as possible. The passively acquired antibodies bind to the toxin, which can no longer bind to host cells or disrupt metabolic processes. The toxin-antibody complexes are usually destroyed by phagocytosis.
Passive immunisation is also used for some serious viral diseases such as rabies, where the antiserum is administered after exposure to the infected bite, together with active vaccination (see Table 2). Passively administered antibodies can also be important for individuals who suffer from certain immunodeficiencies in which they cannot manufacture their own antibodies. Such people are extremely susceptible to many types of infection and administration of pooled human antibodies (which contain a mixture of protective antibodies against many pathogens) can keep them in health. As Table 2 shows, some antisera for passive immunisation may be raised in horses by injecting them with the antigen and later using their serum as a source of specific antibodies.
Activity 8
What problems would you expect to result from infusing human subjects repeatedly with horse antiserum?
Answer
Horse serum contains proteins unique to horses, which are recognised as ‘non-self’ by the human recipient's immune system. The increasingly powerful immune response to these proteins gradually destroys the horse antibodies, so passive protection from the antiserum declines. More seriously, the recipient's blood capillaries can become blocked by aggregates of human antibodies bound to horse proteins, triggering local inflammation and even kidney failure.
Although passive immunisation is now used infrequently due to the introduction of more effective chemotherapeutic agents and active vaccines, it was critically important in the past. For example, horse antiserum prevented thousands of deaths from tetanus among allied soldiers in World War I, and it can still be life-saving in some conditions.
2.2 Summary of Section 2
-
The majority of present-day vaccination programmes use active vaccines containing pathogen-specific antigens to elicit a protective immune response in the recipient. Repeated vaccination enhances the effectiveness of the antibody response and (for some vaccines) also elicits cell-mediated responses.
-
Passive immunisation uses antibodies raised in human donors or in horses, which are used either to treat certain life-threatening infections as soon as symptoms appear, or prophylactically to prevent infection after a known exposure.
3 Critical antigens and the immune response
3.1 How do vaccines work?
Before we consider vaccine design in more detail, it is necessary to point out that not all immune responses protect the host against the target pathogen. Consider antibody-mediated responses induced by a vaccine. Antigens that induce protective antibodies against pathogens (or their products) are known as critical antigens; so if vaccines are to elicit a protective or therapeutic antibody response they must contain pathogen-specific critical antigens.
The external coat of most viruses is relatively simple, since it contains a limited number of antigens, which can be targeted by the immune system. Polio virus has just three protein components exposed on the surface of its capsid. By contrast, bacteria are structurally more complex and generally induce antibodies against a variety of proteins. For example, purified protein derivative (PPD) is an antigenic preparation derived from mycobacteria, which contains up to 200 different antigens. Nevertheless, there are often a limited number of antigens which determine the pathogenicity of the bacteria. In extreme cases, such as tetanus, the toxin is the only relevant antigen that must be neutralised; the bacterium by itself is not invasive or particularly pathogenic, but the toxin is one of the most toxic substances known.
Activity 9
Explain how antibodies that bind to the critical antigens of a pathogen can protect the host from infection.
Answer
Antibodies are multi-purpose defensive molecules, whose functions include: preventing viruses from invading host cells by binding to their attachment sites; ‘labelling’ pathogens for destruction by cytotoxic cells and phagocytes; cross-linking pathogens into immobilised aggregates; directing the lytic complement pathway onto cellular pathogens; and recruiting other components of the immune response to fight an infection.
The pathogenicity of many bacteria depends on their ability to avoid phagocytosis by actively moving away from phagocytes, or by being encased in an anti-phagocytic capsule. Antibodies can overcome these defences. For example, by binding to critical antigens in the capsule they allow phagocytes to engage the bacteria (opsonisation). Antibodies to flagella and other surface components reduce bacterial motility. Antibodies against bacterial enzymes such as collagenases, which promote the spread of streptococci and staphylococci, can also reduce the ability of these bacteria to invade tissues locally.
The critical antigens in some vaccines also induce cell-mediated immune responses in the recipient, which are particularly important in attacking pathogens that replicate inside host cells, i.e. all viruses and some bacteria, including mycobacteria (which cause TB and leprosy), and certain protoctist parasites (e.g. Toxoplasma gondii; Plasmodium species).
3.2 Summary of Section 3
-
Protective immune responses are directed against critical antigens in the pathogen's structure, or in its products (e.g. toxins, enzymes).
-
Antibodies that bind to critical antigens can neutralise or inhibit bacterial toxins and enzymes, immobilise pathogens, prevent them binding to host cells, and enhance their destruction by phagocytes and cytotoxic cells.
4 Strategies for vaccine production
4.1 Introduction
The type of antigen preparation used in active vaccines varies considerably, depending on the pathogen. For diseases such as tetanus, where a bacterial toxin causes the damage, the toxin is first chemically treated to turn it into a harmless toxoid, which is used as the immunising agent. Where protection is required against the pathogen itself, vaccines are based on whole organisms treated in some way to make them safe, or on complex mixtures of antigens taken from the infectious agent. In this section, we discuss the major production strategies for active vaccines, which conventionally contain one or a combination of:
-
intact killed pathogens;
-
live attenuated pathogens;
-
subcellular fragments or molecules from the pathogen – known as subunit vaccines – either alone or linked in ‘conjugates’ to other molecules.
Table 3 gives some examples of commonly used vaccines in each of these categories. The difficulties that can be encountered in developing an effective vaccine are well illustrated in Section 4.2 of the Cholera Case Study (see Section 4.2, which describes some of the many attempts to produce vaccines of all three types. Sometimes vaccine preparations consist of a mixture of components, as in one of the newer vaccines against cholera, which contains inactivated classical and El Tor bacterial strains and a component of the cholera toxin.
| Vaccine type | Infectious disease | Comments |
|---|---|---|
| killed or inactivated vaccines | polio | Salk vaccine (IPV, see Polio Case Study in Section 1.5) |
| cholera | various combinations of El Tor, classical Inaba and Ogawa serotypes (see Cholera Case Study in Section 4.2) | |
| influenza | strains vary annually | |
| whooping cough | killed Bordetella pertussis | |
| typhoid | killed Salmonella typhi | |
| rabies | various strains with similar protection | |
| live, attenuated vaccines | tuberculosis | Bacillus Calmette Guérin (BCG |
| typhoid | oral attenuated strain (Ty21a) | |
| polio | Sabin oral vaccine (OPV, see Polio Case Study in Section 1.5) | |
| cholera | CVD103-HgR strain with attenuated El Tor strain | |
| measles, mumps and rubella | usually combined in MMR vaccine | |
| yellow fever | single strain, stable for decades | |
| chickenpox | attenuated varicella zoster (Oka strain) | |
| subunit vaccines | tetanus | toxoid |
| diphtheria | toxoid | |
| cholera | toxin A or B subunit (used in combination with killed or attenuated strains) | |
| meningococcal meningitis | Groups A and C surface polysaccharides; or conjugate vaccine (MenC) | |
| typhoid | capsular polysaccharide (Vi) | |
| pneumococcal pneumonia | combination of 23 variant surface polysaccharides | |
| haemophilus influenza | type B capsular polysaccharide; or conjugate Hib vaccine | |
| hepatitis B | surface antigen |
In addition, we will look briefly at a number of new approaches to vaccine design currently in development, including:
-
DNA vaccines containing ‘naked’ DNA encoding specific pathogen antigens;
-
Genetic engineering of genes coding for key pathogen antigens either as subunits, or for cloning into non-pathogenic infectious agents used as ‘gene vectors’ for expression in the vaccine recipient.
4.2 Intact killed pathogens
The first deliberate attempts to create a killed vaccine were made by Louis Pasteur in 1885 (Figure 4). He took samples of brain and spinal cord from rabies-infected rabbits and inactivated the (then unknown) infective agent by drying the preparation, or by chemical treatment with formalin. Although many of Pasteur's vaccines were successful in inducing protective immunity, the methods of preparation often generated vaccines with unacceptably high levels of adverse reactions. For example, the rabies vaccine sometimes induced an autoimmune reaction in the central nervous system (CNS) of immunised subjects. The vaccine included molecules from the rabbit CNS, which led to a breakdown in some of its recipients of the normal tolerance to self-molecules found in the CNS. This occurred in only a small proportion of those immunised, but nevertheless the consequences could be fatal.

Many safe killed vaccines have since been produced using chemically inactivated or heat-killed pathogens, but effectiveness is highly variable. For example, the Salk vaccine (IPV) contains an inactivated preparation of the polio virus, which elicits a strong IgG response and long-lasting protection against paralytic polio; by comparison, killed Vibrio cholerae vaccines have generally produced limited immunity for a much shorter period (see the Polio and Cholera Case Studies).
Click to view the Cholera Case Study.
Activity 10
Both the Salk vaccine against polio and killed whole-cell cholera vaccines induce high levels of IgG antibodies. Why are IgG antibodies less useful against cholera than against polio?
Answer
Polio virus must travel from the gut through the blood to the spinal cord before it can cause paralysis. Since IgG antibodies are the principal serum antibody, they can intercept the virus as it moves from one tissue to the other. In contrast, cholera produces its damage by attaching to cells in the gut epithelium, sometimes invading them, and by the release of enterotoxin. So IgG is of limited value in defence against cholera (IgA is much more important).
4.3 Attenuated pathogens
A major strategy for vaccine production has been the generation of attenuated organisms, which retain their antigenicity, but which have lost their pathogenicity. Generally speaking, attenuated vaccines containing killed organisms are less effective at inducing protective immunity than those using live attenuated strains.
Activity 11
Can you think of reasons why a live attenuated vaccine would be better at inducing an immune response than a killed version of the same pathogen?
Answer
The live organisms persist and reproduce for a period in the recipient, presenting a larger and more long-lasting stimulus to the immune system. Also the attenuated strain lives in the appropriate tissue of the host, so it is presented to the immune system by the correct antigen-presenting cells.
In the earliest attempts, the method of producing attenuated strains was to grow the pathogen in vitro, or in laboratory animals over many generations, repeatedly testing to see whether the evolving strain had lost its pathogenicity. The first and most famous example was the strain of Mycobacterium bovis developed in France by Calmette and Guérin (Bacillus Calmette Guérin, BCG), which has been used since 1923 (see Table 1) as a vaccine against M. tuberculosis. Although data from BCG studies show highly variable levels of protection, this has been one of the most widely used of all vaccines – not least because it is cheap. However, it was only with the publication of the complete genome of M. tuberculosis in 1998 that it became clear exactly what the process of attenuation had done. Early in the development of the BCG strain, the bacteria lost a group of nine genes. Moreover, since the original preparation, different strains of BCG in laboratories in different parts of the world have continued to undergo further genetic diversification. Sequencing studies have also shown that M. bovis is quite closely related to M. tuberculosis over several areas of the genome, which explains why they share critical antigens recognised by the protective immune response.
The rationale for culturing a pathogen in vitro or in a non-human species is that it does not require some of its genes (perhaps for transmission, or spread within the body) and consequently these genes may be lost or mutated, (e.g. pox viruses only appear to require 70 per cent of their genes to grow in mammalian cells). However, this process of attenuation has been described as ‘genetic roulette’, since there is no way of knowing what combination of genes will be lost or mutated and the process will produce different strains each time it is carried out.
The Polio Case Study illustrated another problem inherent in using attenuated strains – pathogenic reversion. The oral vaccine contains all three live polio strains in attenuated forms: although the type 1 strain has 57 mutations and has never reverted to the wild type, type 2 and type 3 each have only two relevant mutations, so they require only two reversions to become pathogenic again – as indeed has occurred on a number of occasions.
Activity 12
In general, it has proved easier to attenuate viruses than bacteria. Can you think of any reasons why this should be so?
Answer
It may be because most viruses are genetically less complex than bacteria and contain only a small number of genes, so a few mutations can result in attenuation of pathogenicity. Also most viruses mutate more quickly, so a variant with useful properties in a vaccine is likely to arise more frequently. Bacteria have a number of DNA repair mechanisms that are lacking in viruses, so they can correct or delete mutations that may otherwise have proved useful in a vaccine. Thus attenuation of pathogenicity in bacteria usually requires much larger genetic changes, but the loss of a segment of bacterial DNA often results in the loss of essential functions for life in addition to those for pathogenicity.
One of the most powerful arguments in favour of genetic engineering in vaccine production is that it can deliberately ‘knock out’ the gene sequences responsible for an organism's pathogenicity. The ability to manipulate pathogen genomes and their products is increasingly important in vaccine design, as we discuss below.
4.4 Subunit vaccines
The risk of pathogenic reversion can be overcome if the vaccine contains only fragments (subunits) of the pathogen, but these must include critical antigens in order to provoke a protective immune response. Relatively few subunit vaccines fulfil these criteria, but they include the inactivated toxins (toxoids) of tetanus and diphtheria which have been in use for many years. A subunit vaccine against whooping cough (Acellular Pertussis Vaccine, APV) is under evaluation. Antigen preparations for use in vaccines have also been made from structural components of certain bacteria and viruses, for example, a surface antigen from the hepatitis B virus, or the coat polysaccharides of Neisseria meningitidis or Haemophilus influenzae.
Since the 1990s, a few highly successful conjugate vaccines have been produced, in which a subunit from the target pathogen is irreversibly bound in a ‘conjugate’ with bacterial proteins. The conjugate elicits a greatly enhanced immune response compared with the subunit alone. Several effective conjugate vaccines against H. influenzae type b (Hib) are already in use. In 1999, the UK was the first country to introduce the conjugate MenC vaccine against meningitis caused by Group C meningococci. The vaccine contains a Group C polysaccharide subunit antigen conjugated with either a harmless variant of diphtheria toxin or the tetanus toxoid. Trials are also underway of a conjugate vaccine against genital herpes which links a herpes virus glycoprotein subunit with lipid A, a component of the Gram-negative bacterial envelope.
The components of subunit vaccines have until recently been extracted and purified from cultures of intact pathogens by conventional biochemical techniques, but there is now increasing research into the genetic engineering of critical antigens. If the genes encoding these antigens can be identified and isolated, they can be inserted into the genomes of harmless bacteria or yeasts (genetic recombination). The expression of these genes can yield commercially useful quantities of pathogen-specific antigens as components of recombinant subunit vaccines, such as the current hepatitis B vaccine; others are being evaluated (e.g. against herpes simplex and human papilloma viruses).
4.5 Vaccines of the future
Other applications of modern molecular biology in vaccine design are also coming into play, including DNA vaccines. The use of ‘naked’ (cell-free) DNA encoding critical antigens, directly injected into muscle, seems at first an improbable strategy, since the DNA does not have the necessary cellular machinery for its expression. Nevertheless, if the DNA has a suitable promoter, sufficient can be taken up by cells of the body to allow transcription and production of enough antigen to induce an immune response. The DNA construct is coated onto gold particles which are shot into the tissue using a gas-pressurised ‘gene gun’ (Figure 5). DNA vaccines have been tested in experimental animals with some success, inducing both antibodies and T-cell mediated immunity; trials of DNA constructs from HIV-1 and hepatitis B virus genomes have begun in human volunteers.
Another technique has been to produce genetically-engineered constructs in which the gene encoding a pathogen-specific critical antigen is transported into the vaccine recipient by a harmless virus or bacterium. These vector vaccines immunise the host against the critical antigen, which is expressed by the ‘gene vector’ (Figure 6).
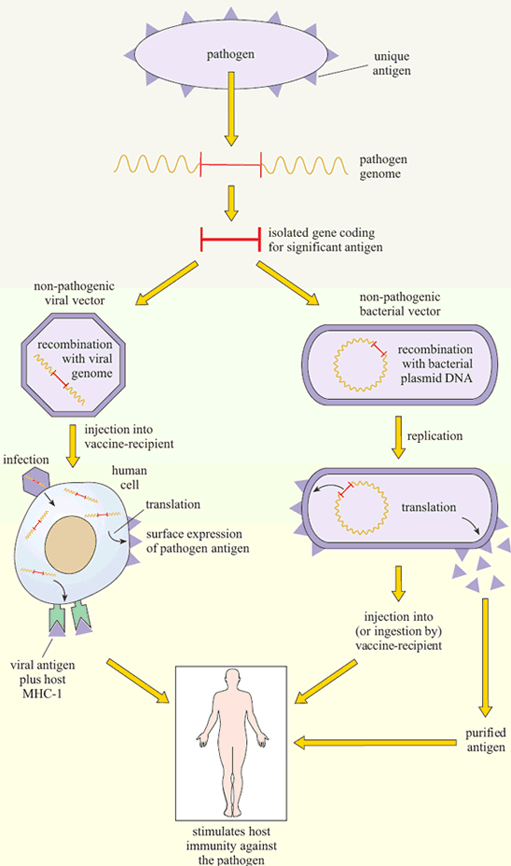
Vaccinia is a popular choice as a gene vector, because it has a long history of relatively safe use in immunisation against smallpox and it can accommodate large amounts of DNA. Moreover, as the use of vaccinia for protection against smallpox has declined since the 1980s, the level of immunity to vaccinia in the population has fallen.
Activity 13
Why is this important in designing a genetically-engineered vaccine assembled in vaccinia viruses?
Answer
If the vaccine recipients are already immune to vaccinia, they would eliminate a vaccinia construct before it could induce immunity to the antigen it was carrying.
Several vector vaccines have been developed to combat HIV infection, so far with limited results. An early version used canarypox virus carrying genes for HIV surface proteins (gp120 and gp41) and encoding some internal molecules including the viral polymerase. A strain of Venezuelan equine encephalitis virus (VEE) has also been used as a vector for gp120. VEE has the advantages of being able to infect human cells and express the HIV glycoprotein, and although it can replicate in human cells, it cannot produce infectious virions. Malaria is another priority target for development of vector vaccines using DNA constructs from Plasmodium carried in vaccinia or fowlpox viruses.
Activity 14
The examples given above all use gene vectors derived from non-human animal viruses: cowpox, canarypox, fowlpox and equine encephalitis. Why are these selected rather than a harmless virus that normally infects humans?
Answer
Firstly, there is the consideration of safety. If a viral vector does not normally infect humans, then it is less likely to revert to a pathogenic type or exchange genetic information with a wild-type human virus. Secondly, recipients will be less likely to have prior immunity to a non-human virus, which would interfere with the development of immunity to the critical antigen in the vaccine.
Bacteria are also under investigation as gene vectors (Figure 6), for example, in the oral typhoid vaccine, Ty21a. There are certain advantages in this approach: bacteria that live in the gut tend to induce strong immune responses in the gut-associated lymphoid tissues (GALT), including a good IgA response. Thus, bacterial vectors that express the antigens they are carrying in the gut would generate the type of immune response required to combat the enteric bacteria and viruses that cause diarrhoeal diseases. At the time of writing (2003) no bacterial constructs have been licensed for mass vaccination in humans, but Ty21 a is being tested as a vector for a variety of bacterial and other antigens, and the first human trial of HIV genes in a bacterial vector has begun.
Finally, an interesting development has been the insertion of single genes encoding critical antigens from bacteria and viruses into plant genomes, including those of potatoes and tomatoes. The antigens are expressed in the plant, which can then be eaten with the aim of inducing an immune response in the gut! The first trial of an oral plant vaccine against hepatitis B virus using genetically engineered potatoes began in the USA in 1999. If it turns out to be protective, it would have a significant effect in developing countries where viral hepatitis is a major health problem. Growing anti-HBV potatoes for local consumption would be a cheap and effective way of protecting the population.
Activity 15
What could limit the efficacy of oral plant vaccines?
Answer
Most proteins in foods are broken down by digestive enzymes in the stomach and intestine, so the critical antigens may be destroyed before they can elicit an immune response. Even if they survive digestion, most people do not usually produce immune responses to antigens in foods, perhaps because food antigens do not induce costimulatory signals for T and B cells.
Figure 7 summarises the current gene-based approaches to vaccine development.
In addition to the pragmatic question of whether a genetically engineered vaccine will induce protective immunity, there is a debate as to whether it is safe to produce such vaccines at all. The arguments are ranged along lines that have been well-rehearsed in more general concerns about genetic engineering. Critics think that altering infectious agents – albeit harmless ones – could produce pathogenic reversion, or further unplanned recombination of the vaccine vector with other bacteria or viruses to produce pathogenic strains. Proponents think that this risk can be made negligible by comprehensively disabling the vectors. The debate is difficult to resolve because although the risks and dangers of contracting an infectious disease are quantified, the risks and dangers from a new genetic vaccine are hypothetical. Other issues of vaccine safety are discussed in Section 8.
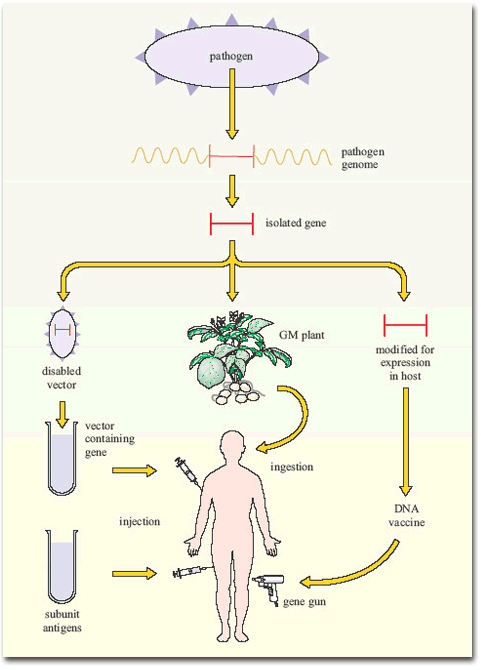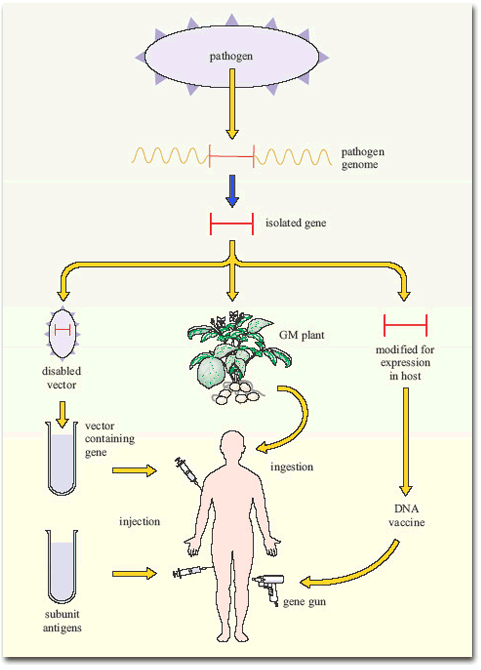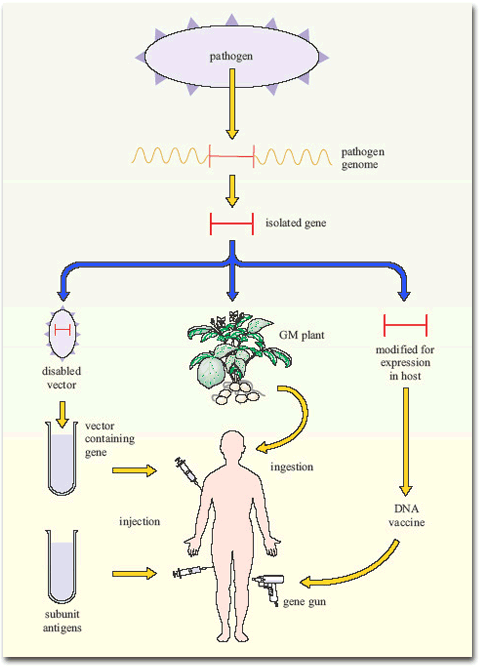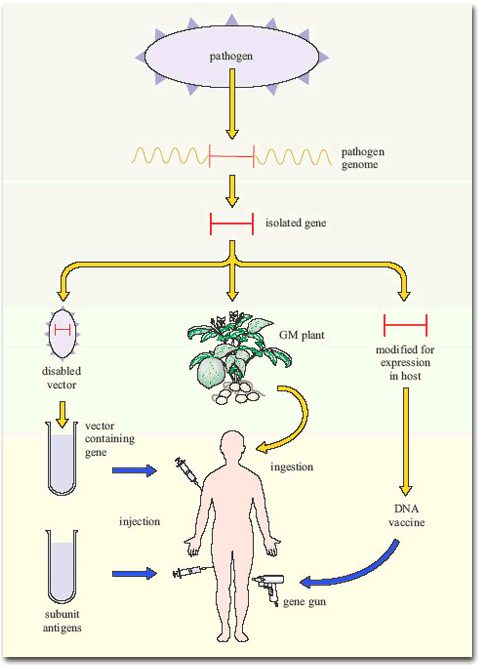
4.6 Summary of Section 4
-
Conventional vaccines contain either killed pathogens or attenuated strains (live or killed) with the same critical antigens as the target pathogen. Genetic manipulation may delete the genes involved in pathogenicity to create new attenuated strains.
-
Subunit vaccines contain pathogen fragments extracted by conventional biochemical techniques, or genetically engineered constructs. The subunit antigen may be conjugated with bacterial proteins or lipids to enhance its ability to induce protective immunity.
-
DNA vaccines contain naked DNA encoding a pathogen-specific antigen which is ‘fired’ into host tissues and expressed there; vector vaccines contain pathogen genes inserted into the genomes of harmless viruses or bacteria. Both vaccine types generate an immune response when the gene product is expressed in the recipient. Oral plant vaccines are genetically engineered to express pathogen antigens with the aim of eliciting protective immunity in the gut when eaten.
5 Influences on vaccine efficacy
5.1 Introduction
The efficacy of a protective vaccine is rarely 100 per cent. Vaccine efficacy can be calculated from the secondary attack rates in vaccinated and unvaccinated individuals and expressed as the relative reduction in the risk of infection in vaccine recipients, compared to the risk in unvaccinated people. In this section, we discuss factors that influence vaccine efficacy and determine whether it reaches the level required to protect a recipient against subsequent infection. We are not concerned here with population effects such as herd immunity and critical immunisation thresholds, which influence the ability of a vaccination programme to eliminate an infection in a community (we consider this in Section 7).
5.2 Antigens and immunogens
In Section 3 we highlighted the need for vaccines to contain pathogen-specific critical antigens, which provoke a protective immune response. Clearly, a vaccine that did not contain critical antigens would be ineffective, but we must now extend the discussion to include other influences on vaccine efficacy. We noted above that pathogens can have antigens that do not induce a protective immune response and that (in most people) antigens in foods do not elicit immunity at all. This introduces an important concept in immunology. It is necessary to distinguish between antigenicity, the ability of a molecule to be recognised as non-self by the cells of the immune system – and immunogenicity, the ability of that antigen to induce an immune response. Immunogenicity is not a fixed property: whether a particular antigen behaves as an immunogen is highly contingent on a number of interacting factors, including:
-
the route by which it is delivered (e.g. orally; injected into the skin; inhaled intranasally); and the quantity of antigen in the ‘dose’;
-
the genetic make-up of the immunised person and how this affects the ability to make an immune response to a particular antigen;
-
the molecular structure of the antigen (e.g. carbohydrate, protein, lipid);
-
the presence of other molecules that enhance immune responsiveness.
Activity 16
How could the route of administration influence the kind of immune response that develops against the antigens in a vaccine?
Answer
Different parts of the body have different antigen-presenting cells, and this determines how an antigen is presented and what kinds of T cells are stimulated.
Antigen presentation partly determines whether a T helper-1 (Th1) or a T helper-2 (Th2) type of response is favoured. Antigens presented to Th1 cells initiate the sequence of events culminating in cell-mediated immune responses; antigens presented to Th2 cells initiate antibody-mediated responses. In practice, both types of response can occur simultaneously, but the route of administration can favour one or the other. It can also influence the class of antibodies that appear: antigens presented in the gut will tend to induce IgA production, since large numbers of IgA-producing B cells are located there; by contrast, antigens injected into the skin will usually be transported to local lymph nodes, where IgG- and IgM-producing B cells predominate.
Activity 17
Explain how the genetic make-up of an individual can affect antigen presentation and why this has an influence on whether a vaccine is immunogenic.
Answer
Antigen-presenting cells take up the vaccine antigens, process them internally and present peptide fragments in the cleft of their own surface MHC class II molecules. The genes encoding the MHC molecules vary between individuals, producing variations in which peptides can be presented to other cells in the immune system. Different individuals will present some protein antigens more efficiently than others. Inefficient antigen-presentation means that these antigens may be less immunogenic in that individual, even though they may provoke a strongly protective immune response in individuals with a different set of MHC class II molecules.
Activity 18
What implication does this have for vaccine design?
Answer
The variation in MHC molecules in a population means that the antigens used in vaccines must be presented efficiently in genetically different individuals.
In addition to the MHC, several other gene loci have been identified that affect the ability to generate a protective immune response; these include genes that affect antigen processing, as well as those involved in cytokine production and cell/cell interactions.
The molecular structure of an antigen can also affect whether it is immunogenic and the type of immune response it generates. Carbohydrates, lipids and glycolipid antigens are processed differently to protein antigens and they are not presented by conventional MHC molecules; for example, glycolipids are presented by a surface molecule designated CD1. Carbohydrates do not generally induce antibody class-switching, so they do not induce the production of high affinity IgG antibodies. We shall not go further into the details of how non-protein antigens are handled by the immune system, but you should note that it is essential that vaccines induce a strong, effective and long-lasting immune response to certain carbohydrates and glycolipids.
Activity 19
Can you explain why?
Answer
The surface of many bacteria consists of carbohydrates and glycolipids, and an effective antibody response against them is highly desirable.
The immunogenicity of pathogen antigens is thus essential to vaccine design, but this is often hard to achieve; for example, some critical antigens are not sufficiently immunogenic in a wide enough range of individuals. However, various other components of the vaccine preparation can be added to enhance the immune response. Such components are called adjuvants.
5.3 Adjuvants
Adjuvants are components of vaccines that enhance their immunogenic potential. In general, they work in one of two ways:
-
They concentrate the antigen in one place (the ‘depot effect’).
-
They activate antigen-presenting cells and induce cytokine production (Figure 8).
The first adjuvants to be devised used antigens in emulsions with aluminium salts, which created depots of antigen and greatly enhanced the levels of antibodies produced in response. Aluminium hydroxide is still added to some vaccines for use in humans, including the diphtheria and tetanus toxoids. Work in animals showed that emulsions containing killed mycobacteria were exceptionally good at producing strong immune responses to other antigens. Mycobacterial products are very effective at activating macrophages and these adjuvants are thought to act by enhancing antigen presentation.
Activity 20
How can bacterial products enhance antigen presentation by macrophages?
Answer
Macrophages have receptors for a variety of bacterial components (including lipopolysaccharide, LPS). Receptor binding to these components causes increased expression of MHC molecules and costimulatory molecules on the macrophage surface, and enhanced secretion of stimulatory cytokines such as IL-1 and TNFa. This upregulation increases the efficiency with which macrophages present antigens to T cells and stimulate T cell activation.
Some bacterial components such as LPS produce adverse local reactions in human recipients, which prevent their use in human vaccines. For example, mycobacteria can cause severe ulceration of the skin, especially in people who have been previously sensitised to them. However, these problems have been overcome in the newer conjugate vaccines (described earlier), which link the antigen irreversibly to bacterial products such as diphtheria or tetanus toxoid, or lipid A. Another technique is to use liposomes, microscopic sacs formed from phospholipids, which trap antigens and ensure that they can be taken up in quantity by antigen-presenting cells. In general, these adjuvants act via the depot effect.
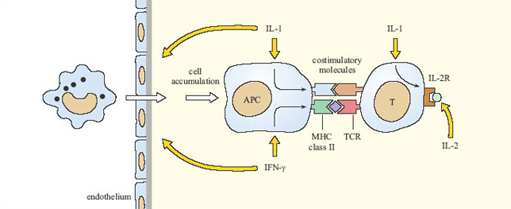
The second category of adjuvants takes a different approach, by incorporating cytokines into the vaccine to enhance the immune response directly. At present (2003) this strategy has been tried in experimental animals, principally using IL-1, IL-2 and IFN-γ, but none have yet been approved for use in vaccines for humans. Figure 8 summarises how these cytokines should (in theory) enhance immune responses to vaccines, although there are many difficulties to be overcome, including the high cost of producing synthetic cytokines.
5.4 Summary of Section 5
-
When designing a vaccine, the antigens it contains must be immunogenic and selected to induce a protective immune response in genetically different recipients.
-
The molecular structure of the antigen, the route of administration and the presence of adjuvants in the vaccine can all influence the efficacy of the immune response, and the type and location of antibody-mediated and cell-mediated defences.
-
Adjuvants in vaccines function either by concentrating antigen in ‘depots’ where they are more effectively taken up by antigen-presenting cells, or by stimulating components of the immune response directly.
6 Challenges to vaccine development
6.1 Introduction
Vaccination seems such a straightforward and effective strategy for controlling infectious disease that the question arises of why we cannot develop vaccines against every pathogen and parasite.
Activity 21
Suggest reasons why effective vaccines already exist (a) against polio, but not against HIV infection; (b) against influenza, but not the common cold; and (c) against tetanus, but not syphilis.
Answer
(a) Polio virus is genetically fairly stable, so the organism does not mutate and there are only three strains to be included in the vaccine. In contrast, the surface molecules of HIV mutate so quickly that antigenic drift occurs continuously even within a single infected individual, making a vaccine extremely difficult to design. And we have yet to discover what would be an effective immune response against HIV.
(b) At any one time there are only a few strains of influenza virus in circulation, so although current vaccines only protect recipients against known strains, the emergence of vaccine-resistant virus is a relatively rare occurrence. Compare this with the viruses that causes colds, of which there are more than 100 in circulation. Moreover influenza is a much more serious illness than the common cold, so there is much more incentive to develop a vaccine against it.
(c) Tetanus presents a well-defined target for the immune response, since we know that antibodies against the toxoid will neutralise it; in contrast we are mostly ignorant of what constitutes an effective immune response against the spirochaetes that cause syphilis.
Comparisons such as these reveal that the factors limiting vaccination strategies to control infectious diseases fall into five broad categories.
-
The nature of the pathogen: including, its distribution in human populations and whether it infects non-human hosts; its mode and speed of transmission; the type of symptoms it causes; whether the disease has a symptom-free latent period or a ‘carrier’ state.
-
The nature of the immune response: including, whether antibody-mediated or cell-mediated immunity predominates in an effective immune response; the type and location of protective responses; the identification and immunogenicity of critical antigens; scientific knowledge of how to enhance the immune response.
-
Economic factors: including, whether it is cost-effective to develop a vaccine and whether a vaccine would be affordable by those who would benefit most from it.
-
Organisational factors: including, whether the infrastructure and personnel necessary to conduct a vaccination programme exist in a susceptible population.
-
Cultural factors: including, the level of public understanding of what vaccination can (and cannot) protect against, and concerns about vaccine safety.
These categories cannot be considered in isolation from each other: the co-evolution of pathogens and their hosts means that (1) and (2) above are intrinsically related and continuously changing; the state of scientific knowledge of the pathogen and host immune responses influence whether (3) the economic case will favour research into a new vaccine; an effective vaccine may be produced, but organisational difficulties (4) may mean that it cannot be delivered where it is most needed (e.g. due to lack of refrigerated storage facilities or trained vaccinators); and (5) concerns about safety may limit vaccine uptake.
In the rest of this section, we consider biological factors of the pathogen and the host, which present challenges to the development of vaccines. In Section 7, we turn to the economic, organisational and cultural limitations on vaccination as a strategy to control infectious disease.
6.2 Zoonoses
Eradication of a disease is a splendid aim, but although vaccines exist against some important zoonotic diseases (e.g. tuberculosis), this goal is extremely difficult or even impossible to achieve for zoonoses. The situation is well illustrated by the viral zoonoses such as yellow fever. An effective vaccine has been available against yellow fever virus for several decades, but the WHO estimates there are still at least 200,000 cases per year with 30,000 deaths (WHO, 2001). The virus is present in monkeys in equatorial Africa and South America and is transmitted from monkeys to humans principally by mosquito bite. It is clearly impossible to eradicate the virus, because there is a very large natural host population of monkeys in which it will persist. Also, the female mosquitoes can transmit the virus ‘vertically’ to their offspring in their eggs, maintaining its presence in the vector population.
Over 200 viruses fall into the category of zoonotic infections (Taylor, 2001), including some of the most lethal human infections ever identified. They include Lassa fever, Hanta virus pulmonary syndrome, Ebola fever and Marburg disease. Fortunately, the incidence of human infection with these diseases is still very low and usually sporadic, but case fatality levels are typically 50–80 per cent (as Figure 9 illustrates). In epidemics, human-to-human transmission readily occurs among close contacts of the original case, but strict quarantine measures have (so far) contained the outbreaks, although complete control has sometimes taken over a year. Nevertheless, the episodic recurrence shown in Figure 9 demonstrates the potential dangers of a large pool of ‘animal’ viruses, either transmitting infection to humans directly or acting as a source for the development of new human viruses.
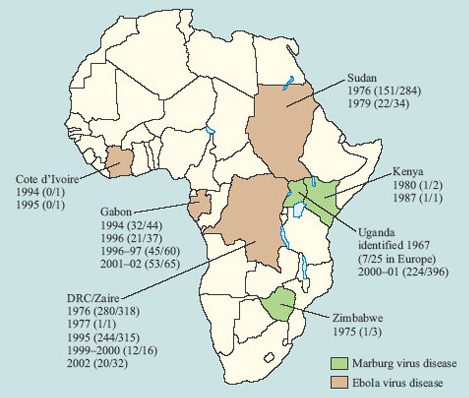
Although it would be desirable to have vaccines against high-fatality viral zoonoses, the animal reservoirs of infection will always be present. Moreover, there are a number of other factors which mean that vaccine development is of relatively low priority. Firstly, if the disease is very rare, the financial incentive for a company to develop a vaccine is lacking. Secondly, there are such a large number of zoonotic viruses that it is difficult to know where to start. An individual who is planning to travel in an area where the infections are found might have to be immunised against a wide range of viruses. Despite these factors, some vaccine development is underway for viral haemorrhagic diseases, since they would be of particular value to health care workers involved in controlling epidemics.
6.3 Asymptomatic carriers
A related problem for disease eradication is seen in some human infectious diseases, where a carrier state develops. One example is typhoid, where 2–5 per cent of individuals become chronically infected, but without any symptoms of the disease. They excrete the typhoid bacteria continuously into their faeces and act as a persistent reservoir of infection. There have been several notable instances of individuals whose work involved food preparation, who have unwittingly passed the disease to dozens of others via the faecal-oral route.
Another example is the hepatitis B virus (HBV), the major cause of progressive liver disease and hepatic cancers worldwide, which can exist in an asymptomatic carrier state. About two billion people worldwide are infected with HBV, of whom 350 million are carriers.Very rarely, health workers in the UK are discovered to be carriers of HBV when they accidentally infect patients. (Medical students in the UK and people who come into contact with human tissue are usually vaccinated against HBV.) The majority of cases of HBV infection in Western countries are now either sexually transmitted or related to intravenous drug-use; the virus can also be transmitted vertically and post-natal transmission from mother to baby is the principal route in South-East Asia.
6.4 Evasion of the immune response
All the infectious agents that cause disease in humans have had to evolve ways of evading our immune responses. Even the zoonoses which have other hosts, and diseases such as tetanus and cholera with causative agents that live in the environment in a free-living form, have adapted to survive for at least a period in human hosts.
Some pathogens mutate or change their surface molecules so rapidly that they keep ‘one step ahead’ of the immune response, at least for a time; some disguise themselves from recognition by the immune system; and others have developed ways of limiting the effectiveness of the mechanisms directed against them.
Activity 22
Give examples of pathogens that evade immune responses by: (a) antigenic variation; (b) antigenic disguise; and (c) countermeasures against immune effector mechanisms.
Answer
(a) Influenza virus and HIV, malarial parasites and trypanosomes all undergo rapid antigenic variation. (b) Schistosomes ‘cloak’ themselves with host proteins, disguising their own surface antigens; herpes viruses and pox viruses appear to have incorporated host genes into their own genomes, enabling them to produce proteins that inactivate the complement lytic pathway, (c) Mycobacteria synthesise proteins that inhibit fusion between the lysosomes containing destructive oxygen intermediates and the phagosomes in which they enter the host cell; staphylococci and streptococci have receptors for antibody Fc regions, which compete with Fc receptors on macrophages – the bacteria ‘trap’ the antibodies so they cannot opsonise the bacteria for destruction by the macrophages.
Some additional points about antigenic variation are worth considering in the context of vaccine design. One of the greatest challenges to vaccine development comes from pathogens that mutate their surface antigens very rapidly. In many cases, the areas that mutate are those on exposed loops of external proteins. In HIV, for example, mutation of specific parts of the pathogen's structure can contribute to evasion of the immune response without interfering with the structural integrity of the virus. There are specific regions on gp120, the large surface glycoprotein of HIV, which are particular targets for antibody responses (i.e. they are immunogenic). These regions are particularly susceptible to mutation, so their shapes ‘drift’ as the antibody response builds up, and new clones of B cells have to be activated to cope with the change. However, these immunogenic regions are not vital functional areas of the gp120 molecule. As the gp120 molecule is required by the virus for attachment to CD4 on helper T cells and macrophages, it cannot mutate randomly since it must always retain its ability to bind to CD4. It is notable that the areas that mutate the most are outside the CD4-binding site.
A different kind of problem is seen in trypanosomes, which switch their variant surface glycoproteins. The sole function of these molecules is to protect the outer surface of the parasite and deflect the antibody response. Although immune responses are effective for a time against one VSG, they are ultimately ineffective in controlling the progression of the disease. There are invariant proteins on the trypanosome surface, but these are much less prevalent and less immunogenic than the VSGs, so they are useless as components of a vaccine. One of the main aims of vaccine designers, therefore, is to induce immune responses to those segments of critical antigens that are constrained and cannot mutate without the pathogen losing a key function.
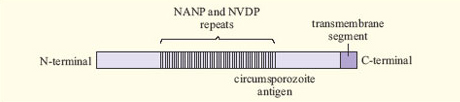
A related problem arises with malaria. The antigens of Plasmodium are extremely complex and vary between different stages of the life cycle of the parasite. Some proteins vary between different Plasmodium strains, while others are relatively constant. An example is the circumsporozoite (CS) antigen, which is involved in attachment of the Plasmodium sporozoite to host liver cells. More than half of the CS protein consists of simple repeats of four amino acids (Figure 10). Such an area may be immunogenic, while being unimportant for protein function.
Activity 23
Explain how other ‘decoy’ proteins of the malaria parasite protect it from host antibody-mediated responses and why they present a challenge for vaccine design.
Answer
Several proteins act as decoys by detaching from the parasite's surface or from the surface of infected red cells. Antibodies against these proteins do not direct an immune response against the parasite itself, and they are ‘mopped up’ by binding to the decoy proteins. Including these proteins in a vaccine would induce the production of antibodies that were similarly ineffective.
The existence of all these escape mechanisms means that it takes a considerable time for even partial immunity to malaria to develop in a naturally-infected population. Consequently, it has been difficult to identify exactly which immune responses would be effective against the parasite, and what to include in a vaccine that could stimulate protective response mechanisms.
6.5 Multiple strains
A similar problem for vaccine design comes from those pathogens that exist in a very large number of different ‘strains’ which are in circulation concurrently. The individual strains are generally stable, so it is not the case (as with rapid mutation and antigenic drift) that new strains are arising all the time, but the numbers of different strains that would need to be included in a vaccine is immense. For example, the streptococci that cause disease in humans can be classified into 20 main serotypes and more than 100 subtypes, based on recognition by specific antibodies. Salmonella bacteria exist in over 2200 different serotypes! This makes it very difficult to design a vaccine that can induce protection against even a significant number of these strains. Apart from the basic problem of identifying and culturing the relevant strains, if too many are included in the vaccine, then the amount of each is too low to induce an adequate immune response. At present, satisfactory vaccines exist for only a few of the pathogenic salmonellae or streptococcal bacteria.
6.6 Prion diseases
Finally, we should look at the particular problems associated with prion diseases, or transmissible spongiform encephalopathies, such as variant Creutzfeld Jacob Disease (vCJD) and kuru. Prion diseases are caused by altered forms (prions) of small membrane glycoproteins, which are normally expressed on a number of cell types in the body, including neurons and lymphocytes. Once prions have appeared, the endogenous protein becomes folded in the same conformation as the prion, so the prions appear to ‘replicate’ and act like an infectious agent.
Activity 24
Why will it be very difficult to develop a vaccine against prion diseases?
Answer
The prion protein has the same primary amino-acid structure as the endogenous protein, so it is perceived as a ‘self-molecule’ by T cells and an immune response cannot develop.
In theory, the conformational differences between the prion and the endogenous protein might allow it to be recognised by B cells and antibodies, because they recognise the overall shape (conformation) of an antigen. However, in practice any differences seem to be too small to be immunogenic, possibly since the B cells do not receive help from T cells.
6.7 Conclusion
Where pathogens show great antigenic variation or multiple strains, the problems of vaccine development revolve around difficulties in identifying critical antigens that show little variation and which induce protective immunity. The challenge to achieve this has not yet been met for a number of important infectious diseases, which still lack effective vaccines (Table 4). Nevertheless, with the possible exception of prions, there are no theoretical reasons why vaccines cannot be developed to give protection against most infectious diseases. However, the limitations on vaccination strategies extend beyond the challenges posed by incomplete biological knowledge – as the final section of this chapter illustrates
| Pathogen | Examples | Disease | Problem with vaccine design |
|---|---|---|---|
| helminths | Schistosoma species | schistosomiasis | antigenic disguise with host proteins |
| protoctists | Plasmodium species | malaria | antigenic variation and morphological complexity |
| Trypanosoma species | sleeping sickness | extreme antigenic variation | |
| fungi | Pneumocystis | fungal pneumonia | ignorance of effective immunity |
| Candida | thrush | ignorance of effective immunity | |
| bacteria | Streptococci | skin and throat infections | multiple serotypes |
| Treponem a pallidum | syphilis | ignorance of effective immunity | |
| viruses | HIV | AIDS | antigenic variation |
| ‘cold’ viruses | common cold | many different types of unrelated virus | |
| prions | vCJD prions | variant Creutzfeldt-Jakob disease | lack of immunogenicity |
†Many other infectious diseases can only be partially controlled by vaccines with low efficacy (e.g. cholera).
6.8 Summary of Section 6
Features of host-pathogen interactions that present a major challenge to vaccine development include:
-
A permanent reservoir of infection in other animals (zoonoses) or in carrier individuals, and free-living pathogens in the environment.
-
Rapid mutation of pathogen genomes resulting in antigenic variation, antigenic disguise with host proteins, multiple strains, or different antigens at different stages of the pathogen's life cycle.
-
Effective countermeasures in pathogens to evade or inhibit immune responses against them including decoy proteins, and inhibition of lysosome fusion complement activation or antigen presentation.
-
Immunogenic regions located away from regions that are essential for pathogen survival and replication.
-
Weak or absent immunogenicity of pathogen antigens, which also creates scientific uncertainty about what constitutes an effective immune response.
7 Limitations on vaccination programmes
7.1 Introduction
Where vaccination programmes have been successful, they have been immensely powerful and effective ways of combating infectious disease. Following the eradication of smallpox, a number of other diseases, including polio, mumps and rubella (German measles) are in line for global eradication over the next 10–20 years. Vaccines could, in theory, be developed for a larger range of infectious diseases than is currently the case, but there are complex economic, organisational and cultural limitations on vaccination programmes that are not easy to disentangle. Some of the major reasons why some diseases are apparently ‘neglected’ as candidates for vaccine control are discussed in this final section; you will readily identify other limitations on vaccination programmes when you conduct an Internet search into their progress at the end of this free course.
7.2 Cost-effectiveness
The lack of a vaccine against certain infectious agents may not be because the scientific knowledge of the pathogen or the host is inadequate, but because it would not be cost-effective to develop one. At first sight, this may seem surprising, given that most vaccines are generally very cheap and effective. The Global Alliance for Vaccines and Immunisations (GAVI, a consortium of international health agencies, charities, governments and pharmaceutical companies formed in 1999) supplies the DTP vaccine against diphtheria, tetanus and pertussis (whooping cough), the oral polio vaccine, the combined MMR measles, mumps and rubella vaccine, and the BCG vaccine against tuberculosis, to immunise children in developing countries at a cost of under US$1 per dose. In addition, there is the cost of organising, conducting and evaluating a mass vaccination programme. Nevertheless, the cost per person for the most widely used vaccines is small – particularly when set against the estimated two million children who still die each year from vaccine-preventable diseases.
However, not all vaccines are cheap: some are priced above the means of the countries that need them most and supplies may not be sufficient to meet demand. For example, 12 million doses of a vaccine against all four of the major strains of meningococcal bacteria are sold to rich countries every year at differential prices, ranging up to US$50 a dose in the USA. An offer by the manufacturers to supply 2 million doses to African countries at $2.75 each was more than they could afford and far below the 10 million doses required (Lancet Infectious Diseases Newsdesk, 2002).
The major expense comes in the initial stages of laboratory research to develop a new vaccine, and particularly in conducting the clinical trials required to establish whether it is safe and effective (Box 1). This cost may be over US$150 million when all expenses are included. Commercial companies take strategic decisions as to whether such investment can be justified in relation to the returns available. The urgent work to develop HIV vaccines has been driven partly by the fact that an estimated 1.5 million people in the USA and Western Europe were living with HIV infection by the end of 2001, causing over 27 000 deaths annually. (Global and regional HIV/AIDS data are regularly updated on the WHO websites.) Given that over 90 per cent of AIDS deaths occur in poor countries, the extent to which HIV affects the rich countries of the world, where citizens would pay a lot for a protective vaccine, has been a factor in the race to develop the first one. But the world market of 6 billion people (since everyone is at risk from HIV) is the greatest possible incentive to investors, and hundreds of millions of dollars have been committed to research into protective and therapeutic HIV vaccines. (For reports on progress, see links to the International AIDS Vaccine Initiative website under Course Resources.)
Box 1 Clinical trials of vaccines
After extensive laboratory tests on animals, clinical trials of vaccine efficacy and safety take place in humans in three phases. Phase 1 trials assess the tolerability of the vaccine in a small number of healthy volunteers. Phase 2 trials test various vaccination regimens (dosage, spacing) for efficacy and tolerability in well-defined groups of a few hundred individuals who are at high risk of infection (e.g. in the case of HIV vaccine trials, the recipients have included injecting-drug users and sex workers). Effects of the vaccine on their immune responses are evaluated and the subsequent rate of infection in the vaccinees is compared with that in control groups who received placebo (dummy) vaccinations, or the existing vaccine where the trial is of a newer preparation. Phase 2 trials of therapeutic vaccines may also be conducted on already-infected subjects, to see if the vaccine slows the progression of the disease or has adverse effects (e.g. promoting a damaging inflammatory response). Phase 3 trials involve large groups of uninfected people (usually thousands) and may have a ‘multi-centre’ design, with different research teams evaluating the outcomes in different locations (see Figure 11). During the Phase 3 trials, attempts will be made to get the vaccine registered for use in the countries in which the first vaccination programme is to be launched. Thereafter, the performance of the vaccine is monitored in mass vaccination programmes in the target populations.

The prospect of lucrative markets in high-income countries has undoubtedly driven research into protective vaccines against human papilloma virus infection, a major contributory factor in cervical cancers, and into the development of a therapeutic vaccine that directs the immune system to destroy the ‘plaques and tangles’ in the brains of people with Alzheimer's disease. By comparison, resources for developing vaccines against parasite-mediated diseases, such as Chagas’ disease and sleeping sickness, which only affect poorer countries, have been comparatively meagre. In some cases the veterinary significance of a zoonotic pathogen or parasite may be given higher priority and this can initiate research into a vaccine that translates subsequently into a preparation for human usage.
Governments, charities and non-commercial organisations also take decisions on the relative merits of vaccines compared with other, more cost-effective, control measures. For example, syphilis is readily treated by antibiotics and the spirochaete has not become antibiotic resistant. Since most antibiotics are also cheap and even easier to administer than vaccines, there have been few incentives to develop a vaccine against syphilis. A similar argument has applied to many other bacterial diseases, such as streptococcal infections and gonorrhoea. However, with the increase in antibiotic resistance in many important bacterial pathogens, the balance of the argument is now shifting towards vaccine development.
For some infectious diseases – particularly the diarrhoeal diseases and those caused by parasites – public health strategies may be as (or more) effective than vaccination. For example, the provision of clean drinking water and sanitation has done more to prevent epidemics of cholera than has any vaccination programme.
7.3 Organisational difficulties
As the Polio Case Study illustrated, the attempt to eradicate a major infectious disease requires an immense effort to organise systematic vaccination programmes throughout all endemic regions, backed up by vigilance to identify residual areas where the pathogen may be persisting or could have been re-introduced. Mass vaccination programmes present a huge logistical task for those who are engaged in their organisation and delivery. Consider what must be involved in administering the National Immunisation Days (NIDs), which aim to vaccinate several million children concurrently at hundreds of centres throughout countries such as India, Somalia or Peru.
Activity 25
Suggest some of the challenges such an event poses for its organisers.
Answer
The inaccessibility of many parts of the country is a major problem, both for getting the vaccination clinics set up in mountainous regions, deserts, tropical rainforests, etc., and for the population who must make long and difficult journeys to attend them. Advertising an NID and explaining its purpose and importance is not straightforward in remote populations with high levels of illiteracy and many spoken languages. Staff must be trained to administer the vaccine correctly and safely; transport and storage facilities for supplies must be organised to ensure adequate population coverage. House-to-house follow-up has to be made to ensure that vaccinations are repeated to ‘boost’ immunity to protective levels.
Organisational problems such as these mean that the conduct of mass vaccination programmes is often less than ideal, particularly but not exclusively in developing countries. For example, the WHO's Immunization Safety Project ( reported in 1998 that up to one third of vaccinations were not being carried out in a way that guaranteed sterility, and only one third of countries importing vaccines had a monitoring system to detect vaccine-associated adverse events. The procedures for ensuring optimum storage of vaccines and disposal of injection equipment were often inadequate, and the most up-to-date vaccines could not always be afforded or obtained.
Activity 26
What ‘perverse effect’ of vaccination could occur in a community where a poorly organised vaccination programme failed to achieve population coverage at or above the critical immunisation threshold?
Answer
The average age at infection of the unimmunised individuals in the population increases, because their contact rate with sources of infection is reduced by the pool of vaccinated people all around them. If the infection is one that causes more severe symptoms or permanent damage in older individuals (e.g. polio, mumps, hepatitis A virus), then a ‘sub-threshold’ vaccination programme will result in an increase in the proportion of adverse outcomes in those who develop the disease at a later age.
Perverse effects such as these have contributed to public anxiety about the safety of vaccination programmes.
7.4 Vaccine safety
Our views on the safety of vaccines have changed enormously since they were first introduced. Consider variolation, which preceded vaccination for smallpox (Section 1). The high fatality rate from smallpox meant that the 2–3 per cent risk of death associated with variolation was considered acceptable in the seventeenth century. Nowadays however, the risk of adverse reactions is a primary consideration, partly because vaccination is usually performed on people who are well, so there is a requirement for very high safety standards.
The production of vaccines is subject to rigorous quality controls to ensure that every batch delivers the same potency, does not induce harmful autoimmune or inflammatory responses, and is free from harmful contaminants. Nevertheless, accidents have occurred in which pathogens survived in supposedly ‘killed’ whole-cell vaccines and live attenuated strains have occasionally reverted to pathogenicity (as the Polio Case Study illustrated). Vaccines are usually produced by growing pathogenic or attenuated strains in laboratory animals or in tissue cultures derived from their cells, and animal proteins may be added to tissue cultures as nutrients.
Activity 27
What additional challenges does this pose for vaccine safety?
Answer
Quality checks must ensure that no ‘animal’ viruses, immunogenic proteins or DNA from the vaccine donor or growth medium are present in the vaccine.
The new DNA vaccines, the use of viral or bacterial ‘gene vectors’ and the possibility that prions may contaminate vaccines, add further levels of complexity. (New challenges for vaccine quality control are reviewed in a WHO report by Dellepiane et al.,2000.)
Most people tolerate the minor discomfort of an injection and the possibility of local swelling, slight fever or other minor symptoms in the short term following a vaccination, but more extreme or long-lasting side-effects are unacceptable. No medical procedure carries a zero risk and vaccinations are no exception, but in particular groups, such as people with immunodeficiencies, the risk is higher and vaccination with live attenuated strains may be inadvisable. Even when a vaccine has a good safety record, the perception of risk and concerns about adverse reactions can have a major impact on vaccine uptake.
7.5 Perceived risk of vaccination
An important consideration in vaccination programmes is the perceived risk associated with the vaccine. As the incidence of a disease falls (possibly as a result of vaccination), the risk of contracting it also falls. Of course, the risk to individuals from the vaccine remains the same – but what changes is the relative importance of the disease compared with the perceived importance of any adverse effects of the vaccine. There is less incentive to be vaccinated or to have a child vaccinated in a population with a low incidence of the disease, and concerns about possible side-effects of the vaccine can further reduce its uptake.
Activity 28
What consequence might falling vaccine uptake have for the incidence of a vaccine-preventable disease in a population?
Answer
As vaccination coverage falls, the proportion who are susceptible to the infection rises and the level of herd immunity to the pathogen declines. If a source of infection enters the population in conditions in which transmission to susceptible hosts is possible, an outbreak of the disease will occur. If the proportion of immunes in the population has fallen below the critical immunisation threshold (i.e. the proportion of the population who must be immunised for the infection to be eliminated), then – in the absence of a return to high vaccine coverage – the infection will become endemic in the community again.
The perception of risk is a subtle and complex phenomenon. Many studies have shown that people tend to overestimate the chance of suffering a rare adverse event (such as being struck by lightning) and underestimate ‘everyday’ sources of risk (such as traffic accidents), which are actually much more likely to happen. Public perception of the risks associated with vaccinations seems to follow this pattern, in that people in countries where a vaccine-preventable disease has fallen to a very low level tend to overestimate the residual risk associated with the vaccine. An additional consideration is that most vaccine recipients are babies or very young children who cannot ‘consent’ to the procedure. Parents may be more reluctant to have children vaccinated than a rational assessment of the risks and benefits would dictate, because they will feel responsible for any adverse outcomes of a procedure they deliberately chose to accept. Two examples of vaccination programmes illustrate these ideas.
In 1999, the new conjugate ‘MenC’ vaccine against Group C Neisseria meningitidis, one of several causes of meningitis, was introduced in the UK in the first mass vaccination programme with this vaccine in the world. An earlier subunit vaccine against Groups A and C was available, but had never been used in a routine vaccination schedule. No vaccine currently protects against Group B, which is twice as prevalent as Group C, but causes a less severe illness. Before 1999, there were about 1500 cases of Group C meningitis in the UK annually, with a 10 per cent case fatality rate and an incidence of serious permanent disabilities in about 15 per cent of those who survived. The septicaemia can cause such rapid and extensive tissue damage in the extremities that amputation of the hands, forearms and lower limbs may be unavoidable.
The vaccine was offered initially to young children, then to everyone of school age, and later to young adults (e.g. college students), these being the highest risk groups. The programme was strongly promoted by the government, health professionals and independent bodies such as the Meningitis Trust. Even though meningitis C is a rare condition – around 5 cases annually per 100,000 children aged under 5 years – parents saw it as such a serious disease that there was a high uptake of the new vaccine. The perceived threat from the disease was considered to be so great that almost all parents accepted government assurances that any risk associated with the new vaccine was low (as indeed turned out to be the case). Following the introduction of the MenC vaccine, the incidence of Group C meningitis fell by about 75 per cent.
Activity 29
Estimate the annual number of lives saved by the introduction of the MenC vaccine in the UK.
Answer
Around 110, based on the figures given above. With 150 fatalities annually, a 75 per cent reduction in the disease incidence implies that mortality is reduced accordingly.
Contrast the lack of public anxiety about vaccine safety in the MenC programme, with the continuing controversy about vaccination against Bordetella pertussis, the causative agent of whooping cough. A pertussis vaccine has been available in the UK since the mid-1950s, accelerating the downward trend in disease incidence. However, in the mid-1970s, concern was expressed about possible neurological consequences of the vaccine. Uptake fell very rapidily from 81 per cent to 30 per cent, followed soon after by a sharp increase in the incidence of whooping cough (Figure 12).
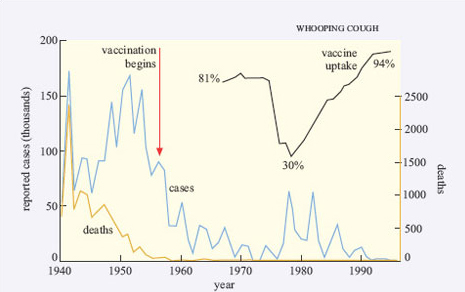
As the perceived threat of the disease rose, parental confidence in the vaccine was re-established and vaccination rates increased again, bringing the incidence of whooping cough back down to almost zero in the early 1990s.
By comparison with bacterial meningitis, whooping cough is highly contagious. The estimated value of R0 for England and Wales in the 1970s, assuming a totally susceptible population, is 16–18. With a transmission risk of 50–90 per cent (i.e. effective transmission between an infective case and a susceptible individual occurs in 50–90 per cent of contacts), most children in an unimmunised population will become infected. Thus, as vaccination rates fell in the 1970s, epidemics very quickly occurred and parents became increasingly concerned about its effects. Historically in the UK, whooping cough was a major cause of infant death, and it still carries a risk of neurological damage or detached retina due to the coughing in a minority of cases. However, as vaccination levels have risen, parental experience of the effects of whooping cough have faded, and concerns about possible adverse reactions to the vaccine have resurfaced.
The cyclical resurgence of whooping cough associated with the decline in vaccination levels highlights the societal dimension of attempts to eradicate an infectious disease. In 2002, concerns were expressed in the UK and in the USA that the combined measles, mumps and rubella vaccine (MMR) might be associated with the development of autism or gastrointestinal abnormalities. Vaccination rates have fallen in those countries, and not just in relation to MMR. Some parents have lobbied for separate vaccines against measles, mumps and rubella to replace the triple vaccine in the belief that this approach would be safer, despite government assertions that it will result in less protection. By the time you are reading this, the debate will have moved on.
7.6 Summary of Section 7
-
The cost of developing and testing a new vaccine is considerable; there is no incentive to develop a vaccine if there are more cost-effective or accessible ways of controlling the disease, if the population at risk cannot pay the market price, or if the disease is so rare that a vaccine would not be profitable.
-
Organisational difficulties in the delivery of mass vaccination programmes can limit their efficacy and (in certain cases) lead to perverse effects, such as an increase in the proportion of adverse outcomes if sub-threshold vaccine coverage increases the average age at infection.
-
Quality control procedures on vaccines must be rigorously applied to ensure that they deliver an effective dose and are not contaminated by potentially harmful material.
-
Perceptions of risk associated with a vaccine can lead to a decline in its uptake and subsequent resurgence of the disease. The level of concern about the safety of a vaccine tends to fluctuate in an inverse relationship with the perceived level of threat from the disease it controls. As disease incidence declines, the safety of the vaccine becomes an overriding consideration.
8 Internet researches into vaccination issues
Conclude your study by undertaking some broad Internet searches on key topics raised in this course. First, bring yourself up to date with the current status of the MMR debate. Spend about one hour on this exercise.
Activity 30
You will certainly find a number of sites with highly polarised opinions in the MMR debate. You should aim to evaluate the reliability of the sites you visit. What evidence do they present? Do they reference their evidence? How much use is made of quotations from individuals? What links do the websites provide? Are links to other sites selective to one side of the debate, or do they address the other side too? Here are some of the other questions that you might consider:
-
Has a fall in the uptake of MMR in any country been associated with an increase in the incidence of measles, or mumps, or rubella? Is it possible to quantify the effect?
-
What evidence has emerged either to support or contradict concerns about a link between MMR and autism or other adverse outcomes?
-
What are the effects of measles in unvaccinated children in developing countries?
-
Have concerns been expressed about possible adverse outcomes of any other vaccine, and (if so) on what evidence has this been based?
Activity 31
Now investigate the details of at least one other vaccination programme (in addition to the polio eradication campaign, which you should already have researched as part of the Polio Case Study). Spend about one hour on this exercise.
-
Identify the type of vaccine used, the method of delivery and any issues that have emerged during the organisation of the programme.
-
What has been the effect over time on the incidence or severity of the disease? How has this varied in different locations?
-
What are the prospects for eradicating the infectious agent globally through mass vaccination programmes? What factors are delaying or threatening progress?
We have provided some links below to websites dedicated to vaccination data and progress reports in the UK, USA and internationally, but there are many others.
Tuberculosis
World Health Organisation fact sheet on tuberculosis.
This site provides a link to the work of STOP TB department and includes the WHO Report 2005 “Global Tuberculosis Control” (full report and summary; links to previous annual Global TB Control reports).
The portal to factsheets and epidemiological data on TB in England and Wales collated by the Health Protection Agency.
The portal to factsheets and epidemiological data on TB collated by the Centers for Disease Control and Prevention (CDC), located in the USA but also collates international data.
Syphilis
The National Institute of Allergy and Infectious Diseases fact sheet on syphilis.
Centre for Disease Control and Prevention (CDC) Elimination of syphilis.
Health Protection Agency. STIs and HIV reports and publications.
Republic of South Africa Department of Health survey report on HIV and syphilis.
GeneQuiz links to information about the T. Pallidum genome. Information on the genomes from other bacteria can be accessed from their home page.
Cholera
This has some FAQ on cholera and info on the cholera Global task Force.
FAQ on cholera from the CDC.
The life and times of Dr John Snow.
A case study of the 1991 peru outbreak.
HIV and AIDs
A tutorial on HIV/AIDS.
Statistics on numbers affected and affected groups.
A comprehensive site on all aspects of HIV.
CDC national prevention network.
A site on treatment. This page has a lot on vaccines.
Polio
The WHO maintains a regularly updated website on the history and progress of the eradication campaign, and the current global status of polio, and links to specific country data and a "Polio News" bulletin.
The UNICEF website on polio focuses on the effects on children.
Information on the biology of picornaviruses, with detailed discussion of polio virus structure, functions and genome.
Polio is the subject of a themed issue of the Bulletin of the World Health Organisation (2000), which contains articles on what makes polio a 'suitable target' for eradication, details of the Angola outbreak of 1999 (the largest ever in Africa), the eradication campaign in specific countries and a consideration of what happens after vaccination stops.
Conclusion
This free course provided an introduction to studying Science. It took you through a series of exercises designed to develop your approach to study and learning at a distance and helped to improve your confidence as an independent learner.
References
Acknowledgements
The content acknowledged below is Proprietary (see terms and conditions) and is used under licence.
Grateful acknowledgement is made to the following sources for permission to reproduce material in this course:
Course image: Apotek Hjartat in Flickr made available under Creative Commons Attribution 2.0 Licence.
Figure 1 Edward Jenner Museum;
Figure 2 Private Collection/Bridgeman;
Figure 3 Roitt, I. et al. (1998) ‘Effect of vaccination on the incidence of viral disease’, Immunology, 5th edn, Elsevier Science Limited.;
Figure 4 Science Photo Library;
Figure 5 Bio-Rad Laboratories;
Figure 9 Reprinted from Armstrong, D., and Cohen, J. (1999) Infectious Diseases, Elsevier Science Limited;
Figure 11 WHO/TDR/M. D. Gupte.
Figure 1.1 Courtesy of George Eastman House;
Figure 1.2 The World Health Organisation;
Figure 1.3 de Quadros, C.A. (1979) ‘Global eradication of poliomyelitis’, Annals of Internal Medicine, Vol. 127, issue 2, pp. 156-158, American College of Physicians;
Figure 2.2 Reprinted from Infectious Diseases by Armstrong and Cohen, Vol 1, p.22.3 by permission of the publisher Mosby;
Figure 3.1 Dr Alan Cann;
Figure 3.2 Reproduced by permission of Jean-Yves Sgro;
Figure 5.2 Photograph provided courtesy of The World Health Organisation.
Don't miss out:
If reading this text has inspired you to learn more, you may be interested in joining the millions of people who discover our free learning resources and qualifications by visiting The Open University - www.open.edu/ openlearn/ free-courses