4 Glucose metabolism: an example of integration of signalling pathways
4.1 Glucose metabolism
We are now in a position to draw together the major concepts and components of signalling, and show how they operate in one well-understood system, namely the regulation of the storage or release of glucose in the human body. From this, you will be able to recognize archetypal pathways represented in specific examples, you will be able to appreciate how the same basic pathways can be stimulated by different hormones in different tissues, and you will see how opposing hormones activate separate pathways that affect the same targets but in opposite ways.
Following a meal, insulin is released into the bloodstream by pancreatic β cells. The overall systemic effects of insulin are to increase uptake of blood glucose into cells, and to promote its storage as glycogen in muscle and liver cells. (Note that glycogen is a polysaccharide consisting of repeated units of glucose used for shortterm energy storage by animal cells.) A rise in the concentration of blood glucose, such as that following the consumption of food, stimulates insulin production, which signals through the insulin RTK. The insulin RTK phosphorylates various substrate proteins, which link to several key signalling pathways such as the Ras–MAP kinase pathway. There are, however, two major pathways that control glycogen synthesis and breakdown in animal cells (Figure 47).
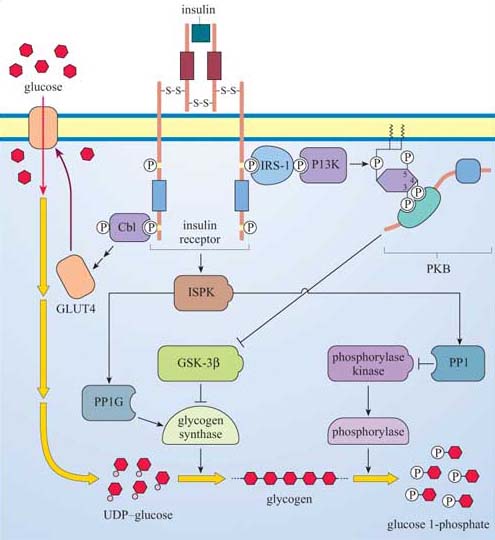
Glygogen synthase kinase-3β (GSK-3β) inhibits glycogen synthase (GS), which is responsible for synthesizing glycogen from uridine diphosphate (UDP)–glucose.
Phosphorylase kinase, a serine–threonine kinase of the CaM kinase family, which, in turn, activates the enzyme phosphorylase, responsible for cleaving glucose 1-phosphate units from the glycogen chain.
Both regulatory mechanisms are influenced by signalling cascades initiated by the interaction between phosphotyrosine residues on the insulin receptor and two signalling molecules:
The first is Cbl, which was introduced in Section 2.4 as a link between RTKs and ubiquitin in receptor sequestration. In this case, it seems to be the start of a pathway that ends up with translocation of GLUT4 glucose-transporter molecules to the plasma membrane, thus promoting the uptake of glucose into the cell.
The second is IRS-1, insulin receptor substrate-1, which is a large protein that binds several SH2-containing proteins, including PI 3-kinase. As described previously (Section 3.3), PI 3-kinase creates phosphorylated inositol phospholipid docking sites for PH-containing proteins, including PKB. GSK-3β is inhibited by PKB, so that its inhibitory action on glycogen synthase activity is negated.
At the same time, an insulin-stimulated protein kinase (ISPK, activated by insulin by an unknown mechanism) acts on the serine–threonine phosphatase PP1G to further enhance glycogen synthase activity. ISPK also activates another serine–threonine phosphatase, PP1, which negatively regulates the activity of phosphorylase kinase, and, ultimately, phosphorylase.
So insulin promotes uptake of glucose into tissues by mobilizing glucose transporters, and in liver and skeletal muscle, activates glycogen synthesis by modulating the activity of GSK-3β and GS, and inhibits glycogen breakdown via PP1. In other words, insulin induces gluconeogenesis in liver and skeletal muscle.
Two hormones, adrenalin (secreted from the adrenal medulla in anticipation of muscle activity) and glucagon (released from pancreatic α cells when blood sugar is low) have the opposite effect to insulin; that is, they increase the rate at which glycogen is converted to glucose (glycogenolysis; Figure 20). In skeletal muscle, glucose enters the glycolytic pathway to produce ATP, the fuel for muscle contraction. In the liver (which is more responsive to glucagon than to adrenalin), glucose is released into the bloodstream for use by muscle cells. Adrenalin and glucagon act through GPCRs, so their signalling pathways start off quite differently from that of insulin, a RTK. However, they end up (among other things) regulating the activity of the same enzymes involved in glycogen synthesis that insulin itself modulates.
Adrenalin has many effects, but in skeletal muscle it acts through β-adrenergic receptors, which are coupled to Gαs and stimulate adenylyl cyclase activity. This results in cAMP elevation and consequently activation of PKA (Figure 48). PKA activates phosphorylase kinase (see (a) in Figure 48), which in turn activates the enzyme phosphorylase. (Phosphorylase kinase activation is also promoted by ACh release from neuron terminals; Figure 48). PKA also phosphorylates three key proteins to promote glycogen breakdown and inhibit its synthesis as shown by letters (b)–(d) in Figure 48:
(b) The serine–threonine phosphatase PP1, which is inhibited as a result (PP1's action is to dephosphorylate, and therefore inhibit, phosphorylase kinase and phosphorylase). Note that PKA phosphorylates (and inactivates) PP1 on different residues than ISPK (which activates PP1).
(c) PP1 inhibitor, which on phosphorylation is activated.
(d) Glycogen synthase, which on phosphorylation is inactivated.
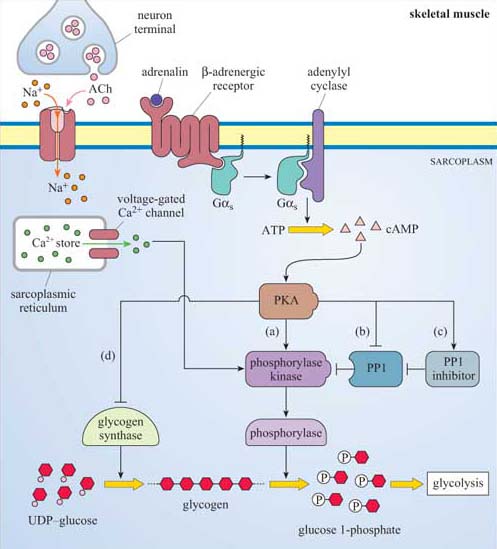
This system provides a good illustration of how a key signalling enzyme (PKA) with multiple substrates can regulate different targets within the same metabolic pathway, all combining to promote one outcome, in this case glycogen breakdown. In at least two cases (glycogen synthase and PP1), it is the same enzymes whose activity is ultimately regulated (either negatively or positively) by insulin (via PKB) and adrenalin (via PKA), acting antagonistically.
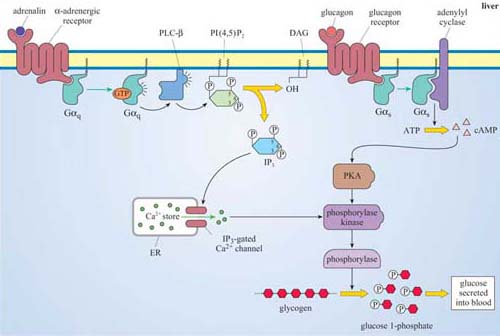
In the liver, however, adrenalin acts mainly but not exclusively through α-adrenergic receptors, which are coupled to Gαq, and activate the PLC/IP3 pathway, which opens IP3-gated Ca2+ channels (Figure 49), helping to activate phophorylase kinase and promote glycogen breakdown, leading to glucose release into the blood.
Glucagon is a hormone that activates glycogen breakdown, particularly in the liver, resulting in a release of glucose into the blood. In the liver, the control of glycogen breakdown is fundamentally the same as in skeletal muscle, but with particular differences. Whereas skeletal muscle needs to be extremely responsive to adrenalin (for the classic fight or flight response), the function of the liver is to maintain blood sugar levels within a constant, physiological range. Liver cells therefore have many glucagon receptors (Figure 49), which are GPCRs coupled to Gαs, activating the same basic pathway that adrenalin does in skeletal muscle, via cAMP/PKA. The result is phosphorylation and activation of phosphorylase kinase, thereby promoting glycogen breakdown.